





El cáncer colorrectal hereditario no polipósico o síndrome de Lynch, causado por mutaciones germinales en genes reparadores de bases desapareadas de ácido desoxirribonucleico (ADN), es la forma más frecuente de cáncer colorrectal hereditario. La identificación de estos individuos no es fácil y se basa en criterios clínicos y moleculares. Se expone a continuación una revisión sobre genética y diagnóstico en el síndrome de Lynch, así como sobre su manejo quirúrgico y prevención.
Hereditary nonpolyposis colorectal cancer or Lynch Syndrome, caused by germinal mutations in mismatch deoxyribonucleic acid (DNA) repair genes, is the most common form of hereditary colorectal cancer. The identification of these individuals is not easy and is based on clinical and molecular criteria. A review is presented on the genetics and diagnosis in Lynch Syndrome, as well as on its surgical management and prevention
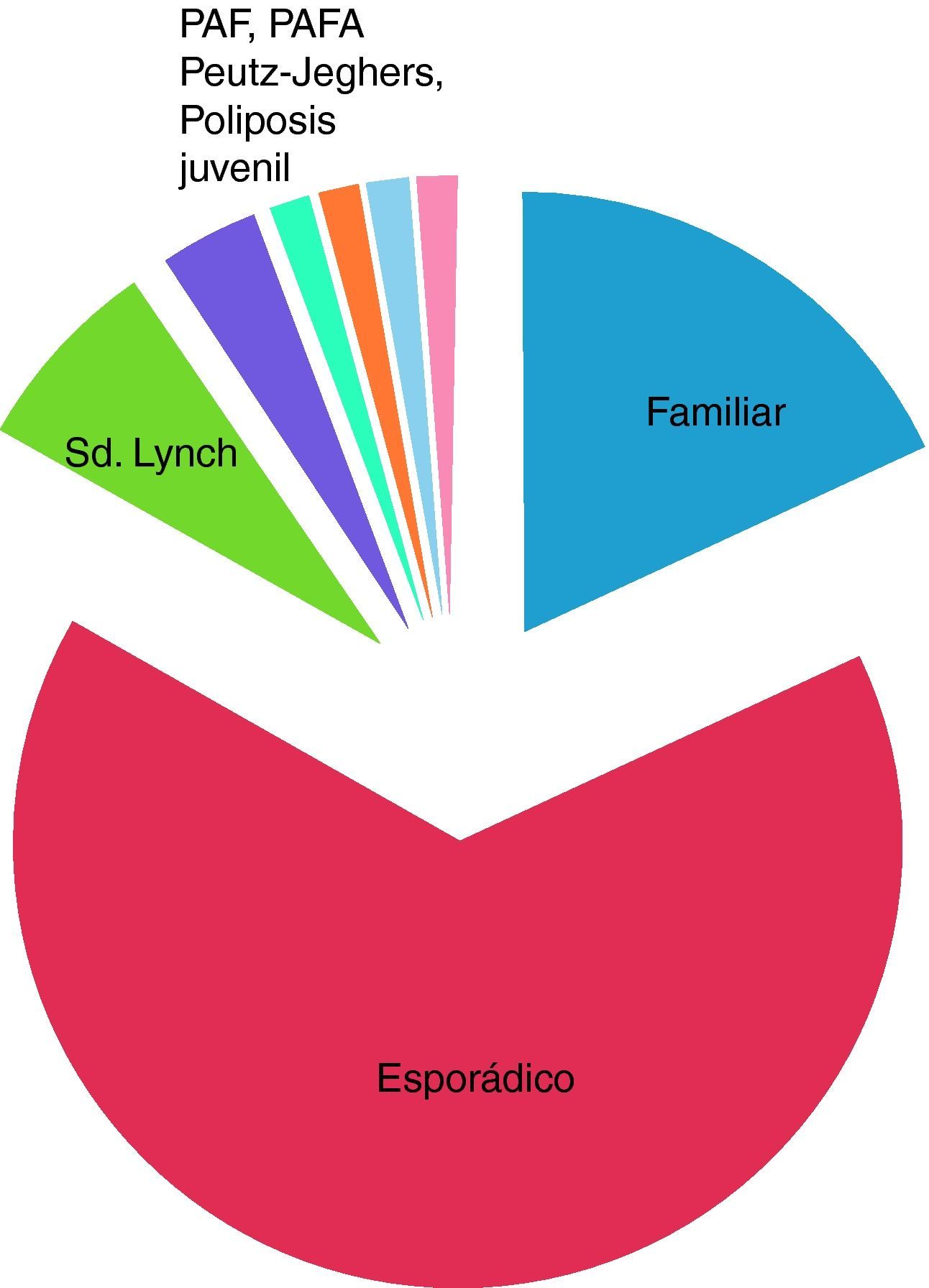
El 80% de los cánceres colorrectales son de aparición esporádica, el 10% son familiares y el restante 5-10%, tienen carácter hereditario1. El cáncer colorrectal con componente hereditario incluye la poliposis adenomatosa familiar y el cáncer colorrectal hereditario no polipósico (CCHNP)2(fig. 1). Preferiblemente se utiliza síndrome de Lynch para denominar a familias en las que se ha identificado una alteración genética en uno de los genes reparadores de los errores de apareamiento de las bases de ADN (Mismatch Repair Genes, MMR). Las principales características del síndrome de Lynch son el desarrollo de cáncer colorrectal a edad temprana (alrededor de los 45 años), la predilección por el colon derecho en más del 70% de los casos, aumento de la incidencia de tumores sincrónicos y metacrónicos, carcinogenia elevada, y un riesgo aumentado de desarrollar neoplasias extracolónicas (endometrio, ovario, gástricas, tracto urinario, intestino delgado, cerebral, hepatobiliar…)3.

Se trata de una enfermedad autosómica dominante debido a la presencia de mutaciones en los genes reparadores de bases desapareadas de ADN, principalmente MSH2 y MLH1, que representan un 90% del total, y con menor frecuencia, MSH6 y PMS22.
Uno de los principales retos en la práctica clínica es la identificación de los individuos portadores de los genes reparadores del ADN, con el fin de favorecer la prevención del cáncer colorrectal a través de oportunas medidas de consejo genético4.
MétodosEste artículo se ha realizado a partir de una revisión de aquellos artículos relevantes en relación al síndrome de Lynch. Dichos artículos han sido identificados a través de una búsqueda bibliográfica en la base de datos MEDLINE, utilizando para ello las siguientes palabras clave: “Lynch Syndrome”, “Nonpolyposis Colorectal Cancer”, “Mismatch Repair Gene”, “Diagnosis”, “Management” y “Screening”. Se han incluido artículos desde septiembre de 1992, hasta marzo de 2010.
Genética y diagnósticoDesde el punto de vista conceptual, hay tres estrategias posibles para identificar a los pacientes con síndrome de Lynch: a) la utilización de criterios clínicos; b) el empleo de técnicas moleculares: inestabilidad de microsatélites (IMS) e inmunohistoquímica (IHQ), y c) la combinación de ambas.
Diferentes criterios se han desarrollado para identificar a familias con síndrome de Lynch. Los criterios de Amsterdam I5 (tabla 1), publicados en el año 1991 fueron fundamentales para establecer una definición de síndrome de Lynch que permitió la identificación de su base genética. Estos criterios sólo tenían en cuenta el riesgo de cáncer colorrectal. Los criterios de Amsterdam II descritos en el año 19996 incluían el riesgo de tumores extracolónicos asociados al síndrome. Dada la baja sensibilidad de los criterios de Amsterdam, y con el reconocimiento de la base genética del síndrome de Lynch, se diseñaron los criterios de Bethesda, recientemente revisados7, con el objetivo de identificar a pacientes con cáncer colorrectal en los que realizar el estudio molecular del tumor para identificar marcadores de deficiencia en la reparación del ADN (IMS) o pérdida de la expresión de la proteína correspondiente al gen mutado por IHQ que sugiera la presencia de una mutación germinal. Estos criterios incluyen la historia familiar y la edad de diagnóstico, pero también las características patológicas sugestivas de inestabilidad microsatelital del tumor (tabla 1).
Criterios de Amsterdam y Bethesda revisados
| Criterios de Amsterdam I |
| Por lo menos tres miembro de la familia con cáncer colorrectal deben estar afectados y deben cumplirse los siguientes criterios: |
| Existe una relación de primer grado entre al menos dos afectos. |
| Al menos dos generaciones consecutiva: están afectadas. |
| Al menos un Cáncer colorrectal se ha diagnosticado antes de los 50 años. |
| La poliposis adenomatosa familiar ha sido excluida. |
| Criterios de Bethesda revisados para testar tumores colorrectales con inestabilidad de microsatélites. |
| Los tumores detectados en el individuo deben ser diagnosticados por IMS en situaciones siguientes: |
| Cáncer colorrectal diagnosticado en pacientes antes de los 50 años. |
| Independientemente de la edad del individuo esiste la presencia de cáncer colorrectal sincrónico, metacrónico u otros tumores asociados a CCHNP. |
| Cáncer colorrectal con H-IMS diagnosticado histológicamente antes de 50 años. |
| Cáncer colorrectal diagnosticado en uno o varios familiares de primer grado afectados con CCHNP o tumores relacionados, y que fueron diagnosticados antes de los 50 años de edad. |
| Cáncer colorrectal diagnosticado en dos o más familiares de primer o de segundo grado e independientemente de la edad. |
Además de los criterios comentados previamente, recientemente han aparecido diferentes modelos matemáticos predictivos para determinar el estado de portador de mutaciones en los genes reparadores de ADN (el modelo de Barnetson8, modelo de PREMM9, modelo MMRpro10).
Los daños del ADN ocurren continuamente en todas las células del organismo, de forma espontánea durante la replicación en la división celular, y como reacción a agentes ambientales. La alteración principal en el desarrollo del síndrome de Lynch consiste en la presencia de mutaciones que afecta a la línea germinal de un grupo de genes muy importantes para el mantenimiento de la estabilidad genómica (MSH2, MLH1, MSH6 y PMS2 entre los más importantes). Estos genes codifican proteínas que están involucradas en el reconocimiento de alteraciones en el apareamiento de bases y en la reparación de estas alteraciones a fin de evitar la aparición de mutaciones. Las alteraciones en este sistema generan una inestabilidad en los microsatélites constituyendo un verdadero marcador de inestabilidad genómica, que se observa en el 90% de casos de CCHNP y en un 15% de casos esporádicos3. Los microsatélites son segmentos muy cortos de ADN (usualmente de 1 a 5 nucleótidos de longitud) que se repiten varias veces a lo largo del genoma. La pérdida de la estabilidad genómica parece ser un paso clave que ocurre en las primeras etapas de la carcinogénesis. Para el estudio de la inestabilidad microsatelital se extrae el ADN del tumor colorrectal y tejido sano en un mismo paciente. La IMS se refiere a los patrones de repetición de los microsatélites que se observa cuando se compara el tamaño del ADN amplificado de tumores frente al de tejido adyacente normal. En ocasiones resulta impreciso establecer la condición IMS empleando este sistema, sobre todo porque no existe un criterio único en cuanto al número de loci que debe ser analizado para diagnosticar la IMS y la proporción de marcadores inestables que debe considerarse para clasificar la IMS. En 1997 se sugirió el empleo de 5 marcadores de IMS para dicho propósito (BAT-25, BAT-26, D2S123, D5S346, D17S250)11. De acuerdo con los criterios internaciones se considera alta frecuencia de IMS (H-IMS) si 2 o más marcadores de los recomendados presentan alteraciones en su secuencia. Se considera baja frecuencia (L-IMS) cuando un solo marcador se encuentra inestable y se considera IMS estable (MMS) cuando ningún marcador presenta variaciones en la longitud de su secuencia. Según algunos autores11 no parece haber diferencias ni patológicas ni clínicas en los tumores colorrectales con L-IMC y MMS. Los tumores H-IMS constituyen el 15% de los cánceres colorrectales y se localizan predominantemente en el colon proximal, presentan características histopatológicas únicas y están asociados con un desarrollo clínico menos agresivo12,13.
Existe una gran variedad de metodologías para la determinación de la IMS. Una de las más empleadas debido a su sencillez y versatilidad es la SSCP (polimorfismo de conformación de cadena simple). Ésta se basa en la relación entre la movilidad electroforética de un segmento de ADN monocatenario y su conformación plegada, la cual a su vez depende estrechamente de la secuencia de nucleótidos o del tamaño del segmento.
Algunas instituciones han introducido el test de IMS como un cribado inicial para cáncer colorrectal, en individuos diagnosticados por debajo de los 50 años o con características histológicas sugerentes de alteraciones de los genes MMR; y aquellos que tienen H-IMS son referidos a estudio genético14.
Ocasionalmente nos podemos encontrar con familias que cumplen los criterios de Amsterdam I, pero en los que no hay ninguna evidencia de IMS, ni detección de mutaciones de los genes reparadores, es decir, que los tumores presentan estabilidad en microsatélites. A estas familias se refiere Lindor et al como “cáncer de colon familiar tipo X”15.
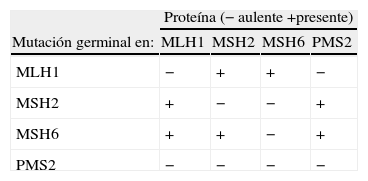
La IHQ es otra de las técnicas empleadas para el diagnóstico de CCHNP, bien complementando al test de IMS o sustituyendo a éste16. En los tumores del síndrome de Lynch puede observarse la pérdida de cualquiera de las proteínas MMR (MLH1 50%, MSH2 39%, MSH6 7%, y PMS2 < 4%). La pérdida de la expresión de MLH1 generalmente se asocia a la pérdida secundaria de PMS2. De la misma forma, la pérdida de MSH2 suele asociarse a la pérdida de MSH6. Sin embargo, las pérdidas aisladas de MSH6 o PMS2, indicativas de mutaciones de estos genes, son poco frecuentes y aisladas13,17. Para valorar el estudio inmonohistoquímico, se dispone de controles internos positivos (estroma, linfocitos) necesarios para poder determinar que la ausencia de expresión en un tumor no es debida a un problema técnico. Un caso se considera con pérdida de expresión cuando no se observa inmunotinción en ninguna célula neoplásica. El resultado debe expresarse como presencia o ausencia de expresión de cada una de las proteínas o como no valorable si no se obtiene controles internos adecuados. La mayoría de los pacientes con mutaciones germinales exhiben en IHQ los resultados mostrados en la tabla 2, aunque pueden aparecen falsos positivos y negativos. La IHQ como método inicial tiene la ventaja de ser una técnica sencilla, barata, además de que permite identificar la proteína no expresada y, por tanto, el gen afecto. Varios estudios han demostrado que utilizando esta técnica para MLH1, MSH2, PMS2 y MSH6, la sensibilidad para el diagnóstico de Lynch es similar a la utilización de IMS con índices superiores al 90%16,18.
Es importante señalar que la inestabilidad microsatelital y la pérdida de la expresión del gen MLH1 también ocurre en un 10-15% de los cánceres colorrectales esporádicos como consecuencia de hipermetilación del promotor de este gen. Para excluir esta posibilidad antes de efectuar el análisis mutacional del gen MLH1 en línea germinal, es útil analizar la presencia de la mutación V600E del gen BRAF en el propio tumor, ya que dicha mutación se asocia a la hipermetilación del promotor del gen MLH1. El test BRAF se puede, por tanto, incorporar al algoritmo del test genético para el síndrome de Lynch previo a la realización del estudio genético19.
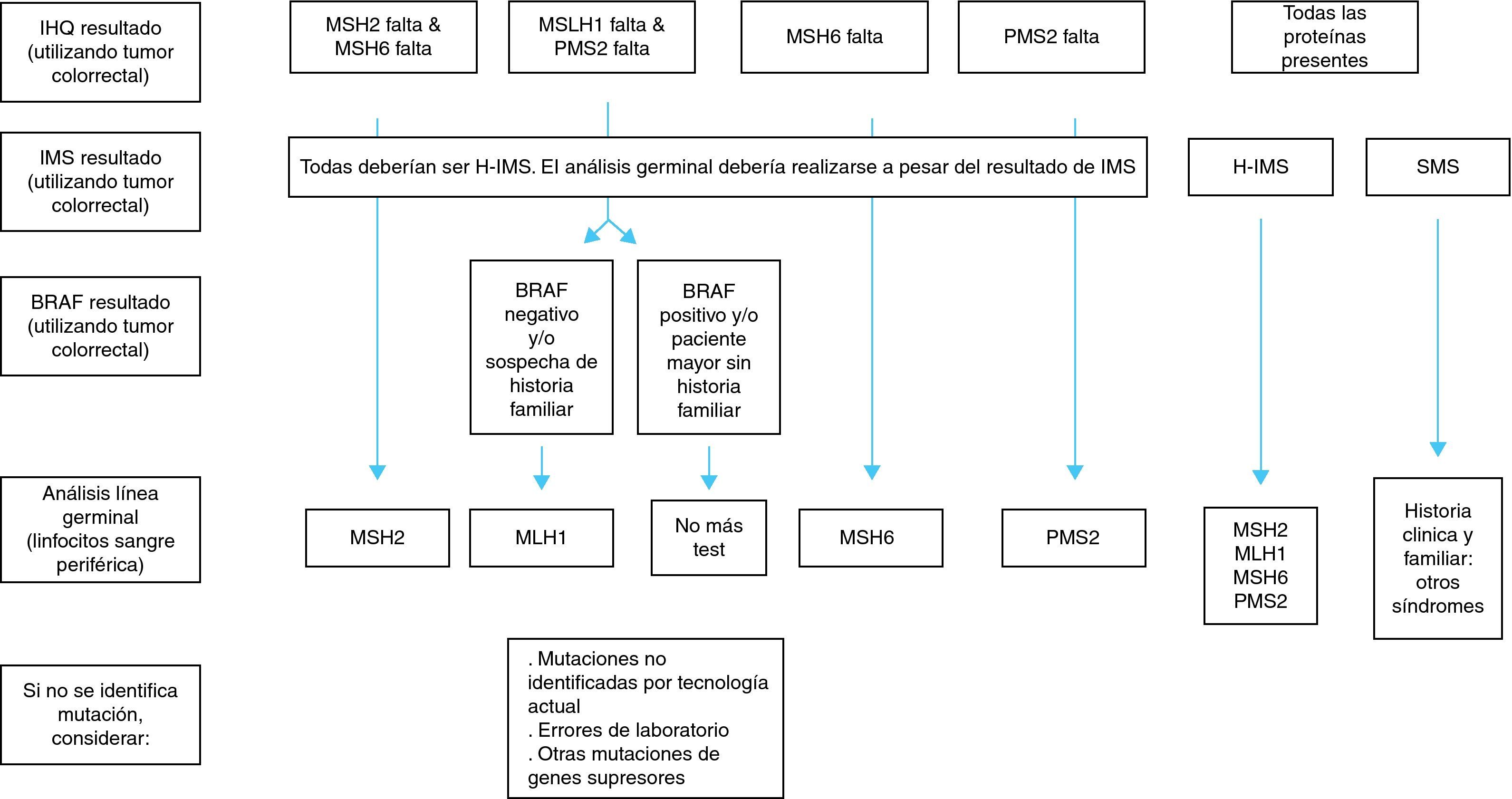
Una vez identificado mediante los criterios clínicos, y utilizando diferentes estudios moleculares (IMS, IHQ, BRAF), se puede continuar con el análisis genético para confirmar el diagnóstico de síndrome de Lynch. Para efectuar el análisis en la línea germinal se analiza el ADN de linfocitos de sangre periférica3,20 (fig. 2).

Según el “European Expert Group”, para aumentar la identificación del cáncer colorrectal hereditario se recomienda: la realización de campañas que informen y conciencien a la población general; en caso de pacientes diagnosticados, será recomendable un estudio familiar completo con el número de familiares afectados, tipo de neoplasia y edad del diagnóstico; guías de referencia con centros genéticos especializados, así como otras que incluyan información referente al cáncer colorrectal familiar y recomendaciones para su seguimiento; considerar la realización de test IMS o IHQ en aquellos pacientes con cáncer colorrectal independientemente de la edad del diagnóstico con un consejo apropiado21.
CribadoUna vez diagnosticado el síndrome de Lynch, es importante su manejo y seguimiento basados en la historia natural del propio síndrome. El cribado del cáncer de colon juega un papel importante, siendo numerosos los estudios que demuestran el beneficio de la colonoscopia en el seguimiento de estos pacientes.
En un estudio en familias con CCHNP, Jarvinen et al demuestran que aquellos pacientes asintomáticos con seguimiento mediante colonoscopia, la incidencia de cáncer colorrectal invasivo se redujo en un 63% en comparación con aquellos de la mismas familias sin seguimiento22. El seguimiento de los mismos pacientes mostró una reducción de la mortalidad en aquellos pacientes con seguimiento y colonoscopia frente a aquellos que no tuvieron seguimiento23. Lindor et al, basados en la evidencia, recomiendan el seguimiento estrecho en los pacientes con síndrome de Lynch, mediante colonoscopia cada 1-2 años, comenzando a la edad de 20-25 años (edad de 30 años en familias con MSH6)24.
Hay una proporción importante de familias que presentan criterios de Amsterdam, pero con resultados de IMS o IHQ negativos. En ellos se recomienda un seguimiento menos intensivo mediante colonoscopia cada 3-5 años a iniciar a los 45 años o bien 5-10 años antes de la edad del diagnóstico de cáncer colorrectal más joven en la familia25.
Siguiendo al cáncer colorrectal, el segundo más frecuente en el síndrome de Lynch es el carcinoma endometrial (40-60% en mujeres con la mutación), seguido del carcinoma de ovario (12-15% de las mujeres con la mutación). Lindor et al, recomiendan ultrasonografía transvaginal y citología anual, comenzando a los 30-35 años24.
La evidencia para el cribado de otros cánceres extracolónicos es limitada. Grupos de expertos de Europa y Norteamérica recomiendan: análisis y citología urinaria cada 1-2 años comenzando a los 30-35 años en aquellos pacientes con antecedentes familiares de neoplasia del tracto urinario; gastroscopia cada 1-2 años comenzando a los 30-35 años para cáncer gástrico en pacientes con antecedentes familiares con este tipo de lesión, o en aquellos que vivan en poblaciones con incidencia elevada de neoplasia gástrica3,24.
Manejo quirúrgico, seguimientoEn aquellos pacientes diagnosticados de Síndrome de Lynch, que desarrollan un cáncer colorrectal, se debe realizar en primer lugar una colonoscopia completa, debido al alto riesgo de presentar un tumor sincrónico. Para la elección de la técnica quirúrgica, se debe tener en cuenta el riesgo de cáncer metacrónico que presentan estos pacientes, que en algunos estudios es del 16% en 10 años26. De ahí que la extensión de la resección se haya debatido durante los últimos años3,27. Aunque no se han llevado a cabo estudios controlados, la mayoría de los expertos defienden la colectomía subtotal con anastomosis ileorrectal3. Von tot Nederveen Cappel et al, comparan la resección colónica segmentaria a la colectomía subtotal en pacientes con cáncer colorrectal diagnosticados de síndrome de Lynch, identificando un aumento en la esperanza de vida en 2,3 años en pacientes jóvenes (< 47 años) a los que se realiza colectomía subtotal. Este estudio se encuentra limitado ya que los datos no se ajustan por calidad de vida, pero el autor sugiere que la resección subtotal puede mejorar la calidad de vida en los pacientes al reducir la necesidad de colonoscopias seriadas, y al disminuir la inquietud de desarrollar una segunda neoplasia28. Un estudio reciente que compara colectomía extendida frente a resecciones limitadas en pacientes con síndrome de Lynch, no muestra diferencias en la supervivencia entre ambos grupos, aunque sí recomienda la realización de colectomía subtotal, ya que el grupo con resección limitada presentan un alto riesgo de desarrollar un segundo cáncer colorrectal y, por tanto, aumenta el número de cirugías abdominales27. La edad del paciente es también un factor a tener en cuenta al decidir la técnica quirúrgica. En pacientes mayores el riesgo de presentar una segunda neoplasia disminuye, y la morbimortalidad de una técnica más agresiva (colectomía subtotal) aumenta29. Es, por tanto, relevante informar al paciente de las diferentes posibilidades técnicas, los riesgos de cada intervención y tomar la decisión en base a los factores del paciente y sus preferencias, con especial énfasis en la edad del mismo y la habilidad para realizar un seguimiento estrecho30,31.
Los pacientes con resecciones limitadas deberán realizar un seguimiento estrecho mediante colonoscopias. Aquellos con resecciones extensas (colectomías subtotales), deberán realizarse proctoscopias seriadas, que son menos invasivas27.
ProfilaxisLa segunda decisión quirúrgica va enfocada a aquellos pacientes con síndrome de Lynch que no han desarrollado un cáncer colorrectal. La cirugía profiláctica evita la necesidad de un seguimiento estrecho endoscópico. Los tipos de cirugía profiláctica disponibles serían la colectomía subtotal con anastomosis ileorectal y la proctocolectomía. La colectomía subtotal obliga a seguimiento postquirúrgico mediante rectoscopias por el riesgo de desarrollar un cáncer rectal metacrónico. Mientras que la mayoría de los cáncer colorrectales en el síndrome de Lynch se desarrollan en el colon derecho, el riesgo de cáncer rectal se estima que es del 11%32. No hay estudios formales que puedan recomendar la práctica de cirugía profiláctica. Aquellos autores que defienden esta cirugía se apoyan en el hecho de que existe un riesgo del 80% para desarrollar cáncer colorrectal en pacientes con CCHNP a lo largo de su vida. Sin embargo, según estudios recientes, debemos tener en cuenta que la penetrancia en el síndrome de Lynch parece ser menor del 80%33 y posiblemente disminuya en pacientes mayores34. Por lo tanto consistiría en realizar una cirugía a un grupo de pacientes que no va a desarrollar cáncer colorrectal a lo largo de su vida, con el riesgo de morbimortalidad operatoria que ésta supone. Parecen necesarios más estudios prospectivos para estimar la penetrancia de cáncer colorrectal en función de la edad en pacientes con síndrome de Lynch y valorar los beneficios de la cirugía profiláctica.
La cirugía profiláctica también se aplica a las neoplasias ginecológicas. La mejor evidencia es un estudio retrospectivo con 315 mujeres con mutaciones en los genes reparadores de las cuales 61 tuvieron cirugía profiláctica. En el seguimiento durante 10 años ninguna de las pacientes con cirugía profiláctica desarrolló cáncer de ovario o endometrio, mientras que en el otro grupo hubo un 33% de cáncer de endometrio y un 5,5% de cáncer de ovario. Estos datos sugieren que la histerectomía y la ooforectomía profiláctica pueden ser una opción razonable en pacientes con síndrome de Lynch que hayan cumplido su deseo de maternidad, discutiendo los riesgos, beneficios y limitaciones del procedimiento24.
Diferentes estudios han demostrado la eficacia de los antiinflamatorios no esteroideos y la aspirina en la reducción de la incidencia de pólipos adenomatosos colorrectales esporádicos y cáncer. Sin embargo, su eficacia entre los pacientes con síndrome de Lynch aun es desconocida24. Los anticonceptivos orales han demostrado una disminución del riesgo de cáncer de ovario y endometrio en la población general. No hay datos que sugieran el beneficio en cánceres extracolónicos entre pacientes con síndrome de Lynch. El efecto de la quimioterapia en pacientes con MSI-H o CCHNP se ha reportado sólo en pequeños estudios. La mayoría muestran que no hay beneficio en el tratamiento con 5-FU en estos pacientes. Hasta la fecha no hay suficiente evidencia para utilizar quimioterapia siendo necesarios ensayos controlados y prospectivos para recomendaciones futuras3,24.
Consejo genéticoEl consejo genético es un proceso que proporciona, tanto al paciente como a los miembros de su familia, información sobre el riesgo de presentar o transmitir a su descendencia una determinada susceptibilidad genética a desarrollar una neoplasia. Informa, además, sobre la posibilidad de hacer un diagnóstico molecular, y sobre cuáles son las medidas disponibles para su prevención y diagnóstico precoz35.
Debemos tener en cuenta el estado emocional del paciente y de la familia, que generalmente requieren apoyo psicológico. Se estima que un enfoque adecuado y detallado requiere al menos 60-90 minutos. La utilización de consejeros genéticos experimentados en el consejo de miembros de una familia con síndrome de Lynch, permite al especialista utilizar su tiempo de manera más eficaz y enfocarlo en el manejo clínico13. Por eso, ante la sospecha clínica de un posible síndrome de Lynch, el paciente y la familia deberían ser derivados para su evaluación a una unidad de cáncer hereditario/consejo genético36.
En el proceso de consejo genético se evaluará el riesgo personal y familiar de susceptibilidad hereditaria al cáncer mediante la realización de una historia exhaustiva sobre los antecedentes personales y familiares, siendo esta historia familiar una pieza clave de la calidad de la estimación del riesgo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

