






El linfedema es una enfermedad progresiva sin curación definitiva conocida. En países desarrollados la incidencia más alta se observa tras cirugía del cáncer de mama y radioterapia. Puede ser clasificado como primario o secundario. El linfedema primario es consecuencia de una alteración de desarrollo del sistema linfático y se ha subdividido en 3 formas: linfedema congénito propiamente dicho, forma precoz y forma tardía (enfermedad de Meige), dependiendo de la edad de presentación. Estas formas son habitualmente esporádicas y generalmente afectan a extremidades inferiores. Un subconjunto de pacientes con linfedema congénito presenta un patrón familiar autosómico y dominante: enfermedad de Milroy1. El linfedema secundario se debe a las consecuencias de cirugía, radiación, infección, invasión tumoral o fenómenos compresivos en los vasos y los ganglios linfáticos o tras quemaduras graves o picaduras de insectos, como se ha descrito. La causa más frecuente a escala mundial de linfedema es la filariasis, que es especialmente común en el sudeste de Asia y África. En Occidente prevalece la etiología posquirúrgica. Las complicaciones del linfedema crónico de extremidades son la celulitis y el linfangiosarcoma, si bien otras neoplasias de naturaleza diferente, tales como el carcinoma epidermoide2, el linfoma3 de células B y angiosarcoma4, se han descrito.
El linfangiosarcoma es un tumor vascular infrecuente que asienta habitualmente sobre linfedema de larga evolución: síndrome de Stewart-Treves, descrito en 1948, con unos 300 casos registrados hasta la actualidad y que aparece al cabo de algunos años tras mastectomía y radioterapia habitualmente. Sucede también en el linfedema congénito o puberal (enfermedades de Milroy y Meige) y en el linfedema no hereditario (congénito, temprano o tardío)5.
Material y métodos
El objetivo fue recoger los casos observados de linfangioma en un hospital intentando analizar sus características: la edad, el sexo, la ubicación, el tratamiento y los datos de seguimiento (es decir, recidiva tumoral y enfermedad metastásica). Dada la rareza del proceso y la limitación numérica existente en este tipo de enfermedad, se ha realizado un análisis descriptivo.
Resultados
Hemos estudiado 5 casos: 3 casos de síndrome de Stewart-Treves tras mastectomía y radioterapia y 2 en pacientes afectados de linfedema congénito (forma tardía). Cuatro eran mujeres y 1 varón. Las edades de los pacientes fueron de 24, 37, 54, 65 y 52 años en el momento del diagnóstico (figs. 1 y 2). El diagnóstico definitivo fue establecido por biopsia en todos ellos. En 3 casos se realizaron técnicas de inmunotinción para antígenos específicos de angiosarcoma (laminina), que resultó positiva en todos ellos. Un paciente presentaba metástasis cuando fue atendido. Los datos generales de los mismos aparecen registrados en la tabla 1. Se realizó cirugía radical en 4 pacientes. Las técnicas empleadas fueron: amputación supragoneal: 1; desarticulación de cadera: 1; desarticulación escapulohumeral: 2, y 1 tratado con gemcitabina y radioterapia ante la existencia de enfermedad metastásica en el momento del diagnóstico. Dentro de los primeros 14 meses de seguimiento fallecieron 3 pacientes y 2 se encuentran libres de enfermedad pasados 46 y 86 meses, respectivamente.
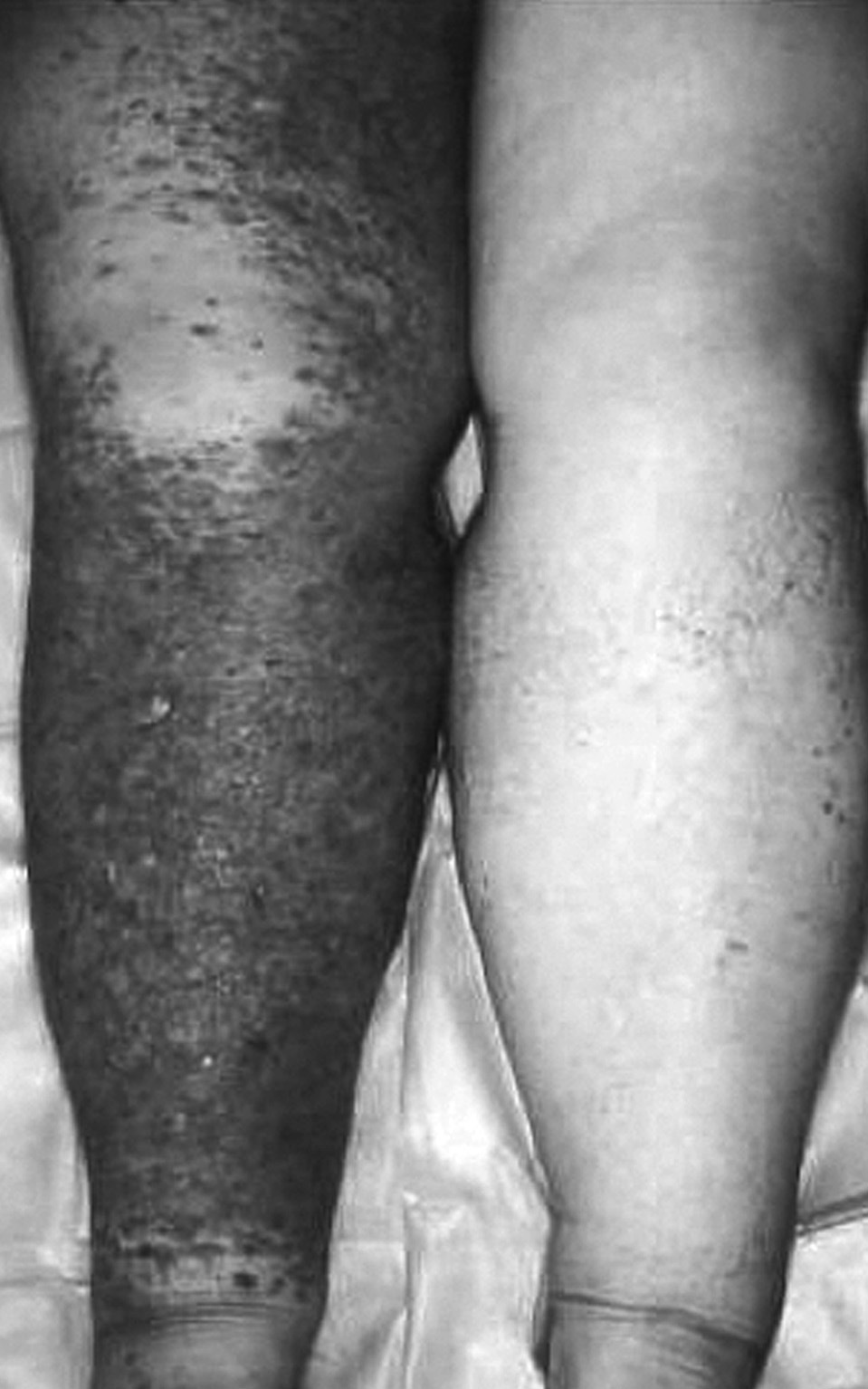
Fig. 1. Caso 3. Linfangiosarcoma sobre linfedema congénito.
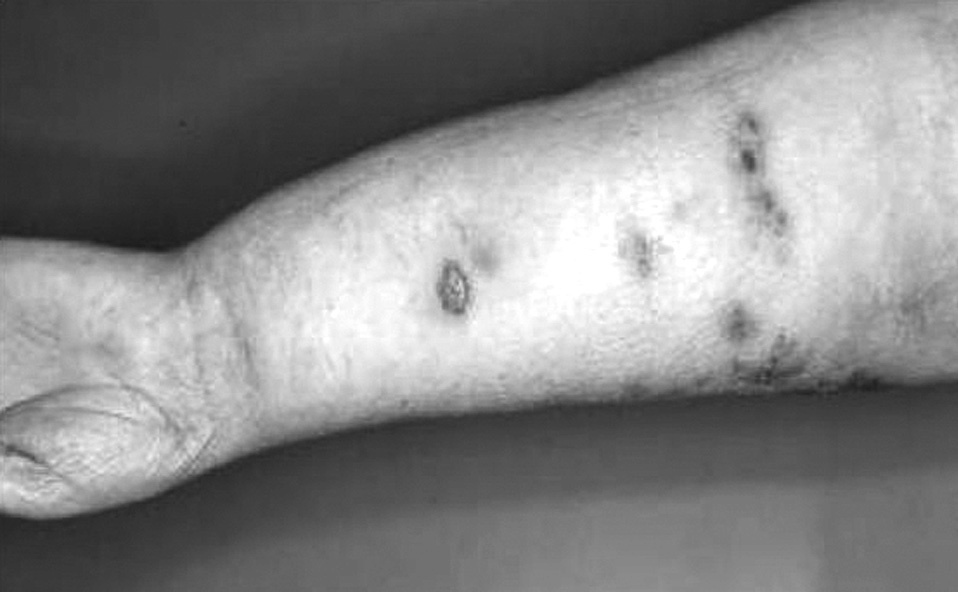
Fig. 2. Caso 4. Síndrome de Stewart-Treves posmastectomía.
Discusión
El linfangiosarcoma es un tumor vascular infrecuente que asienta habitualmente sobre linfedema de larga evolución: síndrome de Stewart-Treves posmastectomía, linfedema congénito o puberal (enfermedades de Milroy y Meige) y en el linfedema no hereditario (congénito, temprano o tardío)5. Las transformaciones malignas son una característica común de estos síndromes.
En 1948, Stewart y Treves6 describieron 6 casos después de mastectomía radical. Este síndrome es una forma excepcional de angiosarcoma que ocurre como una complicación de linfedema. El linfedema y la linfangiectasia crónicos que preceden al linfangiosarcoma pueden ser inducidos por una mastectomía radical con afección ganglionar y terapia postoperatoria de radiación. El linfedema postraumático, congénito o espontáneo crónico, se puede asociar también con linfangiosarcoma. Un intervalo de varios años parece ser necesario para la transformación maligna. Tomita et al7 demostraron que el intervalo entre mastectomía radical y la aparición del linfangiosarcoma oscilaba entre 5 y 14 años. En una serie de 22 pacientes el intervalo entre mastectomía y el comienzo del angiosarcoma era de 11 años (5-16 años). Hay una incidencia elevada de múltiples tumores primarios en la misma extremidad afectada. Generalmente tiene un pronóstico muy grave8.
En 1959, McConnell y Haslam9 dividieron el curso del desarrollo del linfangiosarcoma en 3 etapas.
1. Linfedema de la larga evolución.
2. Fase premaligna o de angiomatosis.
3. Angiosarcoma establecido.
En caso de linfedema de larga evolución o en formas congénitas, la aparición de nódulos violáceos o pápulas así como aparentes equimosis traumáticas deben ser una advertencia diagnóstica. El diámetro externo (> 5 cm), la profundidad de la invasión (> 3 mm), la tasa de mitotis (> 3 HPF), los márgenes, la recidiva tumoral y los márgenes quirúrgicos positivos son elementos pronósticos claramente establecidos. La combinación del síndrome de Maffucci y el de Stewart-Treves se ha descrito con presencia de múltiples encondromas y hemangiomas cutáneos conjuntamente.
La incidencia acumulativa de linfangiosarcoma tras irradiación por cáncer de mama es del 0,2% (intervalo de confianza [IC] del 95%, 0,09-0,47) en 10 años. La proporción estandarizada de la incidencia (SPI) (observados n.° de casos [Obs]/esperados n.o de casos [Esp] del Registro Danés del Cáncer para el período estudiado) resultó de 1,81 (IC del 95%, 0,91-3,23); p = 0,03. El número de personas/años en riesgo (obs-esp)/100.000 durante el mismo período/(100.000) era 9,92. Este estudio sugiere que pacientes tratados por la radiación para el cáncer de mama tienen un riesgo de desarrollar sarcoma más alto que el de la población general10.
Aunque el síndrome de Stewart-Treves se conoce también como linfangiosarcoma, los estudios ultraestructurales e inmunohistológicos muestran que esta malignidad surge de vasos sanguíneos más que de vasos linfáticos. Los anticuerpos contra el factor VIII lo relacionan como marcador de células endoteliales, si bien no se manifiesta habitualmente en las células neoplásicas. Un marcador más sensible en este sentido es la inmunotinción de la lectina.
También el antígeno CD34 es marcador de células endoteliales vasculares y no reacciona con el endotelio linfático. Los marcadores positivos para laminina, CD34, el colágeno IV y la vimentina pueden ayudar a clasificar estos tumores como angiosarcomas.
Estudios de imagen como la MRI se recomiendan para evaluar la extensión local; sin embargo, su valor no lo es tanto cuando se refiere a la definición de límites marginales. Para exclusión de la enfermedad metastásica la tomografía computarizada es el procedimiento de elección.
La biopsia es esencial para el diagnóstico del linfangiosarcoma. La punción-biopsia-aspiración es inadecuada para el diagnóstico en estos casos.
Además, debemos recalcar la importancia del examen clínico regular de todos los pacientes afectados por linfedema crónico. De hecho, aunque el pronóstico de este tumor depende de que el diagnóstico sea precoz y la cirugía exerética, pronta y asociada a radioterapia, puede aumentar la posibilidad de supervivencia de estos pacientes. La amputación o la exéresis local amplia proporcionan la mejor oportunidad de supervivencia a largo plazo en pacientes con síndrome de Stewart-Treves. La quimioterapia con mitoxantrona intraarterial y paclitaxel con prueba de sensibilidad ex vivo previa parece un enfoque complementario, adecuado y actual. Asimismo, la gemcitabina es un agente sumamente activo para el angiosarcoma y el linfangiosarcoma. Algunos autores no encontraron diferencia significativa en las tasas de supervivencia de pacientes tratados con quimioterapia comparadas con los tratados con radioterapia.

