




Autoimmune pancreatitis (AIP)1 is a benign fibroinflammatory disease that frequently presents as obstructive jaundice, which may or may not be associated with a pancreatic mass. It shows characteristic histological changes, and there is excellent response to corticosteroid therapy, as published in the 2011 International Consensus on AIP.2
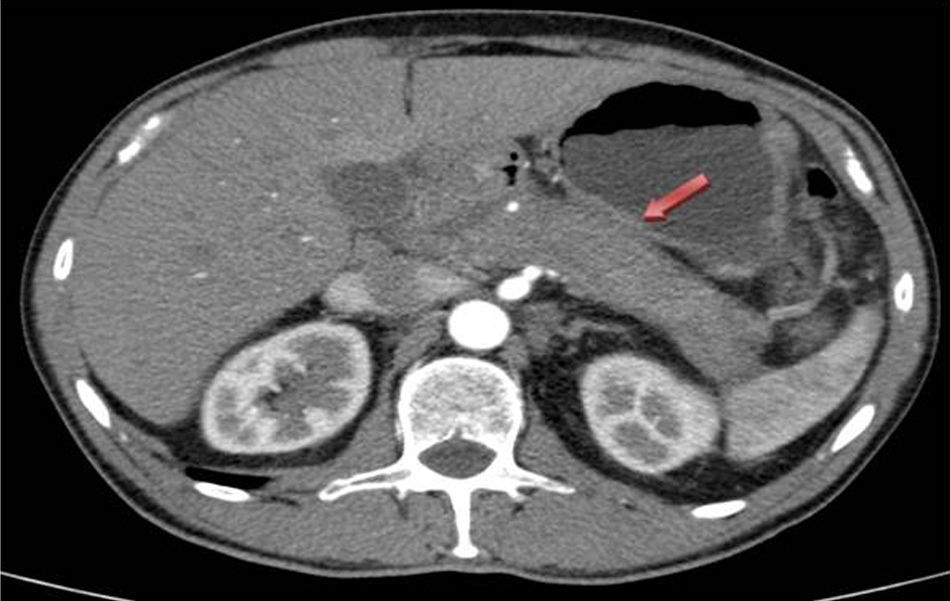
We report the case of a 59-year-old male with no prior history of interest who was transferred to the general surgery department due to symptoms compatible with post-ERCP acute cholecystitis after having been admitted to the Gastrointestinal Department because of obstructive jaundice secondary to a mass in the head of the pancreas. CT scan showed evidence of increased pancreatic gland size, related with acute pancreatitis vs a neoformation, as well as dilatation of the intra and extrahepatic bile duct (Fig. 1). Endoscopic ultrasound showed that the entire pancreatic gland was increased in size, with a neoformation in the head measuring 43×32mm, which was in contact with the superior mesenteric vein by 12mm. The Wirsung duct had a beaded appearance with a clear caliber throughout. The extrahepatic bile duct was dilated and had defined walls, with no interior content, but the distal section was displaced by the previously described mass. ERCP revealed irregular stenosis of the proximal intrapancreatic common bile duct and dilatation of the main bile duct. Lab analyses showed elevated total bilirubin, at the expense of direct bilirubin (3.2mg/dL), normal tumor markers and slightly elevated immunoglobulin G4 169mg/dL (adults: 9–104mg/dL). The pancreatic biopsy taken during endoscopic ultrasound was not conclusive for malignancy.

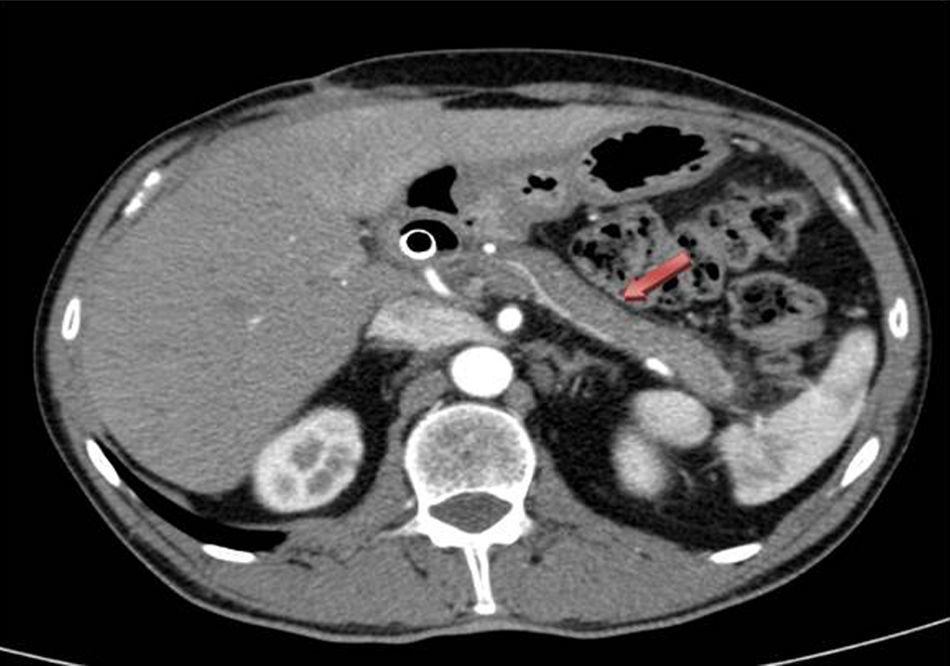
Given the poor evolution of the acute clinical symptoms and the uncertain pancreatic diagnosis, we decided to perform urgent surgery, and a cholecystectomy and pancreatic biopsies were performed. The postoperative period progressed favorably, and the patient was discharged on the 7th day post-op. The pathology results from the pancreatic biopsies were compatible with type 1 AIP because histologically there was extensive fibrosis with an isolated whirl pattern, lymphoplasmacytic aggregates, polyclonal plasma cells, many of which had IgG4 expression in some fields of up to >50 positive cells/high power field. Given these findings, treatment was initiated with corticosteroids, which achieved radiological improvement as seen on CT of the pancreatic inflammatory process (body and tail) one month after the start of treatment (Fig. 2).

AIP is an uncommon disease (prevalence: 2% of chronic pancreatitis) whose symptoms (obstructive jaundice) and radiology results (pancreatic mass or obstructive bile duct lesion)3 are similar to those of pancreatic cancer. This leads to a high percentage of pancreatic surgical resections in a benign disease that otherwise responds well to treatment with corticosteroid therapy.
Histologically, AIP presents well-defined changes that are easily distinguishable from changes that occur in other types of pancreatitis (chronic alcoholic or obstructive), since the lymphoplasmacytic infiltrate is dense and more pronounced around medium and large-sized ducts, compressing the ductal lumen (horseshoe or star duct images are very characteristic of AIP), which differs from the ductal dilatation characteristic of chronic pancreatitis of other origins. Characteristic findings of AIP include lymphoplasmacytic sclerosing pancreatitis (LPSP) without granulocytic lesions; meanwhile, the pathognomonic findings of type 2 AIP are idiopathic duct-centric pancreatitis (IDCP) with granulocytic lesions.4
Currently, there are no specific serological markers for the diagnosis of AIP. Elevated serum levels of IgG4 is a characteristic finding of type 1 AIP (type 2 AIP never presents increased IgG4). Some studies accept the cut point of 135mg/dL for IgG4 values suggestive of AIP versus pancreatic cancer with a sensitivity and specificity of 95 and 97%, respectively.5
In accordance with the International Consensus for the diagnosis of AIP,2 the recommended treatment is prednisone at an initial dose of 35–40mg/day6 or 0.6–1mg/kg/day for 4 weeks. Afterwards, if there is clinical and radiological response, the dose is gradually reduced over 3–4 weeks. Some groups recommend maintaining treatment with corticosteroids at low doses (2.5–5mg/day) for 3 years in type 1 AIP, given its elevated rate of recurrence. The persistence of elevated IgG4 and proximal stenosis of the bile duct after treatment are factors for recurrence. The reintroduction of corticosteroids or the start of immunosuppressants are therapeutic alternatives used in these cases.7
Given the reported evidence of pancreatic adenocarcinoma or intraductal papillary mucinous neoplasm in patients with AIP,8,9 close follow-up of these patients is necessary.
Thanks to the surgical experience accumulated in patients with acute pancreatitis (technical difficulties: vascular injury and hemorrhage10), we propose that in patients with uncertain diagnosis (atypical findings: atypical clinical evolution, involvement of other organs, peripheral halo around the mass, lack of pre-stenotic duct dilatation or the elevation of serum IgG4 to diagnostic levels), it is important to reconsider the situation and assess surgical biopsies prior to pancreaticoduodenectomy or to assess the response to corticosteroids, as long as pancreatic cancer has been ruled out with a high level of certainty (pancreatic masses with an atypical radiological image and negative cytology for malignant cells).
FundingThe authors declare having received no funding to complete this study.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Valero Liñán AS, Rueda Martínez JL, González Masiá JA, Miota de Llama JI, González Masegosa P. ¿Pancreatitis autoinmune o cáncer de páncreas? Cir Esp. 2016;94:415–417.
