




La pancreatitis autoinmune (PAI)1 es una enfermedad fibrinoinflamatoria benigna que se manifiesta frecuentemente como ictericia obstructiva asociada o no a masa pancreática, que cursa con cambios histológicos característicos, y que presenta una respuesta excelente al tratamiento con corticoides, como queda recogido en el Consenso Internacional para la PAI del 20112.
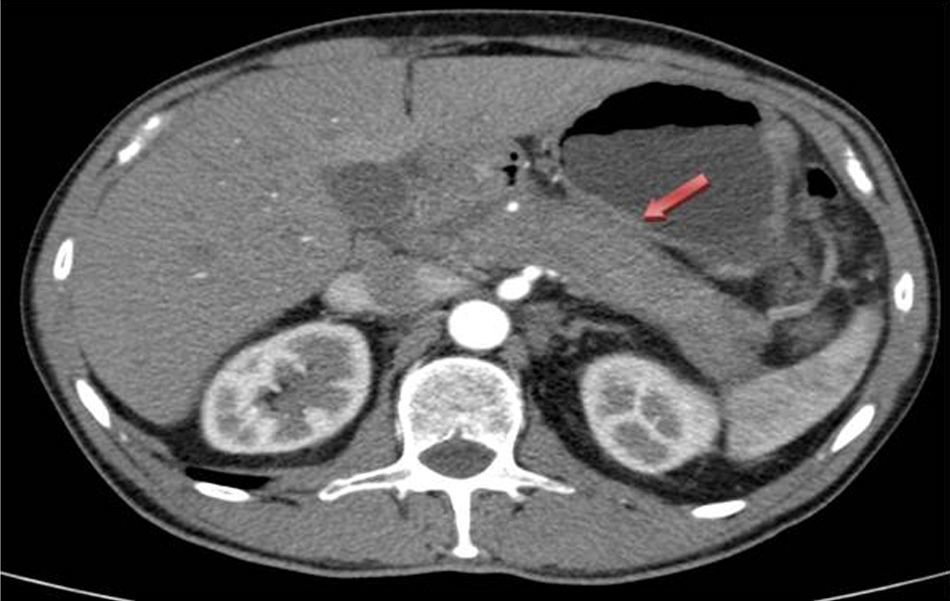
Presentamos el caso de un varón de 59 años de edad, sin antecedentes de interés, que fue trasladado al servicio de cirugía general por cuadro compatible con colecistitis aguda post-CPRE tras ser ingresado en el servicio de digestivo por ictericia obstructiva secundaria a masa en cabeza de páncreas. Se le realizó TAC que evidenció aumento de tamaño de la glándula pancreática, en relación con pancreatitis aguda vs. proceso neoformativo, así como dilatación de la vía biliar intra y extrahepática (fig. 1). La ecoendoscopia mostró páncreas aumentado de tamaño en toda la glándula, con neoformación en cabeza de 43×32mm, que contacta con la vena mesentérica superior en 12mm. El conducto de Wirsung está arrosariado, y con calibre patente en todo el trayecto. La vía biliar extrahepática, dilatada de paredes definidas, sin contenido en su interior, pero el tramo distal está desplazado por la lesión previamente descrita. La CPRE reveló estenosis irregular del colédoco intrapancreático proximal con dilatación de la vía biliar principal. Las pruebas de laboratorio mostraron bilirrubina total elevada, a expensas de la directa (3,2mg/dl), marcadores tumorales normales e inmunoglobulina G4 ligeramente elevada 169mg/dl (adultos: 9-104mg/dl). La biopsia pancreática realizada por ecoendocopia fue no concluyente para malignidad.

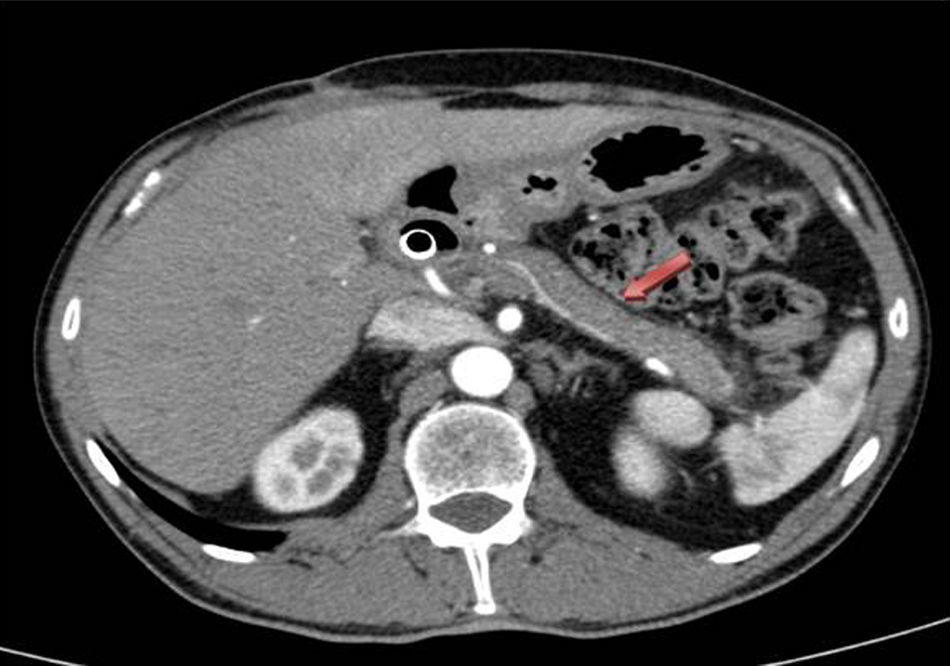
Dada la mala evolución del cuadro clínico agudo, y la duda diagnóstica a nivel pancreático, se decide intervención quirúrgica urgente, realizando colecistectomía más biopsias pancreáticas. El postoperatorio evoluciona favorablemente, por lo que se procedió al alta al 7.° día posquirúrgico. El resultado anatomopatológico de las biopsias de páncreas es compatible a la PAI tipo 1, ya que histológicamente presenta extensa fibrosis con aislado patrón arremolinado, agregados linfoplasmocitarios, células plasmáticas policlonales, muchas con expresión de IgG4 en algún campo hasta >50 células positivas/campo de gran aumento. Ante estos hallazgos se inició tratamiento con corticoides, consiguiendo una mejoría radiológica, por TC, del proceso inflamatorio en páncreas (cuerpo y cola) al mes de iniciar el mismo (fig. 2).

La PAI al ser una enfermedad poco frecuente (prevalencia del 2% de las pancreatitis crónicas), y al manifestarse tanto clínica (ictericia obstructiva) como radiológicamente (masa pancreática o lesión obstructiva de la vía biliar)3 de forma similar al cáncer de páncreas es causa de un elevado porcentaje de resecciones quirúrgicas pancreáticas, por una enfermedad benigna que responde al tratamiento con corticoides.
Histológicamente, la PAI presenta unos cambios bien definidos, que son fácilmente distinguibles de los cambios ocurridos en otros tipos de pancreatitis (crónica alcohólica u obstructiva), ya que el infiltrado linfoplasmocitario es denso y se acentúa en torno a los ductos de mediano y gran tamaño, comprimiendo la luz ductal (imagen ductal en herradura o en estrella, muy característica de la PAI) que difiere de la dilatación ductal característica de la pancreatitis crónica de otro origen. Los hallazgos característicos de la PAI tipo 1 son: pancreatitis esclerosante linfoplasmocitaria (PELP) sin lesiones granulocíticas, mientras que los hallazgos patognomónicos de la PAI tipo 2 son: pancreatitis idiopática ductocéntrica (PIDC) con lesiones granulocíticas4.
En la actualidad no existen marcadores serológicos específicos para el diagnóstico de la PAI. La elevación sérica de IgG4 es un dato característico de la PAI tipo 1 (PAI tipo 2 nunca cursa con aumento de IgG4). Algunos estudios aceptan el punto de corte de 135mg/dl para valores de la IgG4 sugestivos de la PAI frente al cáncer de páncreas con una sensibilidad y especificidad del 95 y del 97%, respectivamente5.
El tratamiento recomendado es prednisona a una dosis inicial de 35-40mg/día6 o 0,6-1mg/kg/día, según el Consenso Internacional para el diagnóstico de la PAI2, durante 4 semanas y, si ha habido respuesta radiológica y clínica, disminuir gradualmente la dosis a lo largo de 3-4 meses. Algunos grupos recomiendan mantener el tratamiento con corticoides a dosis bajas (2,5-5mg/día) durante 3 años en la PAI tipo 1, dada su elevada tasa de recurrencia. La persistencia de IgG4 elevada, así como la estenosis proximal del conducto biliar tras el tratamiento son factores de recurrencia. La reintroducción de los corticoides o el inicio de inmunosupresores son alternativas terapéuticas utilizadas en estos casos7.
Dada la evidencia descrita de casos de adenocarcinoma de páncreas o neoplasia papilar intraductal mucinosa en pacientes con PAI8,9, se hace necesario el seguimiento estrecho de estos pacientes.
Gracias a la experiencia quirúrgica acumulada en pacientes con pancreatitis aguda (dificultades técnicas: lesión vascular y hemorragia10), proponemos que en aquellos pacientes que exista una duda diagnóstica (hallazgos atípicos: evolución clínica atípica, afectación de otros órganos, existencia de halo periférico en la masa, falta de dilatación ductal preestenótica o la elevación a niveles diagnósticos de la IgG4 sérica) es importante replantear la situación y valorar biopsias quirúrgicas previas a la duodenopancreatectomía cefálica o valorar la respuesta a corticoides, siempre y cuando hayamos excluido con un alto índice de seguridad la existencia de cáncer de páncreas (masas pancreáticas con una imagen radiológica atípica y citología negativa para células malignas).
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

