




La poliposis adenomatosa familiar (PAF) es una patología hereditaria, caracterizada por la existencia de pólipos y cáncer en el colon. La PAF puede ser consencuencia de dos trastornos genéticos: El gen adenomatous polyposis coli (APC) o el gen mutación Y homólogo (MUTYH). Las diferencias clínicas y fenotipicas entre las dos alteraciones geneticas no estan claramente establecidas.
Materiales y métodosSe realizó un análisis restrospectivo de las manifestaciones clínicas, criterios quirúrgicos, características histológicas, tipo de mutación y resultados a largo plazo de pacientes diagnósticados mediante análisis genéticos de poliposis adenomatosa familiar entre 1984 y 2018.
ResultadosDe un total de 71 pacientes incluidos en el estudio, en 14 de ellos se identificó mutación del gen MUTYH y en 57, mutación del gen APC. A 60 pacientes se les realizó tratamiento quirúrgico, a la mitad de ellos se les practicó proctocolectomía y a la otra mitad, colectomía total. En pacientes con la mutación APC, el 63% presentó adenomas duodenales; el 61%, pólipos gástricos y el 54% tumor desmoide. De los pacientes con la mutación del gen MUTYH, el 21% presentó adenomas duodenales y al 21% se le diagnosticó pólipos gástricos. En el 21% de los pacientes con mutación del gen APC, el número de pólipos fue inferior a 100 y en el 64% de los pacientes que presentaron mutación del gen MUTYH se observaron más de 100 pólipos en el colon. No existió diferencias estadísticamente significativas entre lo grupos respecto a la proporción de pacientes con más de 100 pólipos.
ConclusiónEs importante valorar la afectación colónica y la extracolónica en pacientes con mutaciones genéticas asociadas a la PAF.
Familial adenomatous polyposis is described as one of the common two types of genetic disorders: APC and MUTYH gene associated polyposis syndrome and the clinical differences between the two can sometimes be unclear.
Materials and methodsA retrospective analysis and comparison was made of clinical, surgical, and histological criteria, mutation types and the long-term results of patients who underwent genetic analysis which resulted in the diagnosis of Familial Adenomatous Polyposis between 1984 and 2018.
ResultsOf the total 71 patients included in the study, 14 were identified with the MUTYH gene, and 57 with the APC mutation. In patients with the APC mutation, 63% had duodenal adenoma, 61% gastric polyp and 54% had desmoid tumor. Of the patients with the MUTYH mutation, 21% had duodenal adenoma and 21% were diagnosed with gastric polyps. In 21% of the patients with APC mutation, the polyp count was <100, and 64% of those with the MUTYH mutation had >100 polyps in the colon No statistical difference was determined between the groups in respect of the proportion of patients with >100 polyps.
ConclusionThe pre-operative genetic testing of patients with polyposis coli will be useful in determining the future clinical outcome and helpful in guiding an informed decision as to whether to apply surgical treatment. It is useful to determine the colonic and extra-colonic involvement of genetic mutation diseases in patients with Familial adenomatous polyposis.
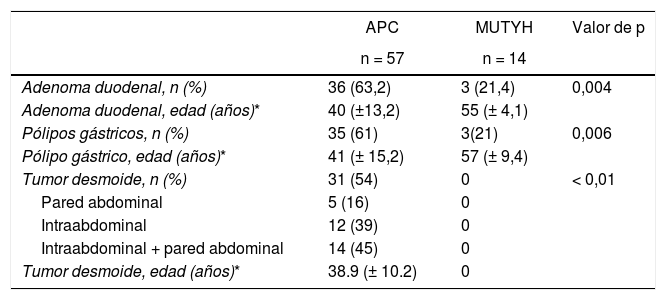
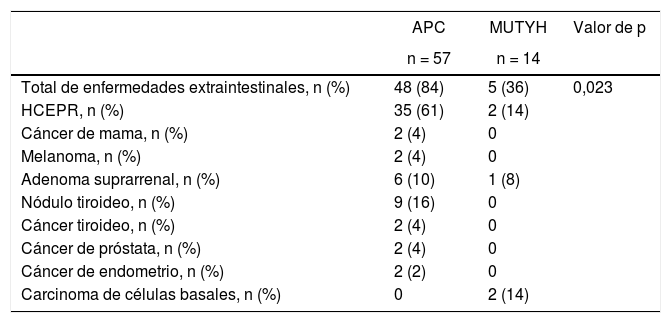
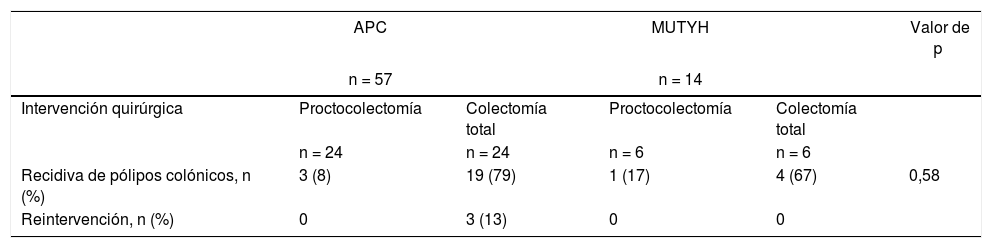
La poliposis adenomatosa familiar tiene una incidencia en la población general de 3-10 casos por 10.000 habitantes1,2. Es una enfermedad de transmisión autosómica dominante y estos rasgos se pueden observar en el linaje familiar3. La causa principal es la mutación del gen adenomatous polyposis coli(APC), que es un gen supresor tumoral ubicado en el cromosoma 5q21-q22. Se han descrito más de 840 mutaciones APC que afectan a una gran cantidad de puntos en el gen, cuya región más afectada es el exón 153,4. La ubicación del área de invasión de la mutación determina los detalles fenotípicos. Otra mutación que rara vez se observa es la poliposis adenomatosa asociada al gen MUTYH (PAM). El gen MUTYH del locus lp34 del cromosoma 1, junto con otros genes, pertenece al sistema de reparación del ADN, conocido como «reparación por escisión de bases»5. Genes como POLE, POLD1, NTHL1 y MSH3 también contribuyen a la etiología del pólipo adenomatoso colorectal6,7.
El objetivo de este estudio fue establecer la extensión y la cronología de la lesión situada tanto en la región colónica como extracólonica y su desenlace clínico a largo plazo en los pacientes con mutaciones genéticas para optimizar el tratamiento de los pacientes diagnósticas de PAF.
Materiales y métodosSe realizó una revisión retrospectiva de pacientes diagnosticados de PAF entre 1984 y 2018 a partir de análisis genéticos. Se analizaron y compararon los resultados clínicos, radiológicos, quirúrgicos y anatomopatológicos, y los datos de seguimiento de los pacientes identificados. Se excluyó del estudio a aquellos pacientes con datos incompletos o no disponibles. El diagnóstico se estableció mediante el análisis genético de todos los pacientes. Respecto a los casos con antecedentes familiares de certeza, el análisis genético se utilizó únicamente cuando no existía un diagnóstico clínico. Posteriormente a estos pacientes, se les realizó una prueba genética de confirmación.
Pruebas genéticasLa extracción de ADN se realizó con muestras de sangre periférica y mediante la secuenciación de la integridad del gen si no se encontraba una mutación en la región del complejo con la mutación (codón 1250-1550) de APC. En los pacientes sin una mutación APC, se realizó una búsqueda de la mutación MUTYH, como se ha descrito con anterioridad.
Según el diagnóstico, los pacientes se dividieron en dos grupos: El grupo 1 estaba formado por pacientes con mutación del gen APC y el grupo 2, por pacientes con la mutación del gen MUTYH. Se compararon la edad y el sexo de los pacientes de los dos grupos, así como el tipo de colectomía si habían sido intervenidos quirúrgicamente, el tiempo de operación y si la cirugía fue laparoscópica o abierta (convencional).
Los pacientes se clasificaron según dos fenotipos: tipo clásico si se encontraron más de 100 pólipos y tipo atenuado si estos eran menos de 100 pólipos (PAFA, poliposis adenomatosa familiar atenuada). Se determinó la localización predominante de los pólipos en el colon. Durante el estudio, también se registró si inicialmente se les había diagnosticado cáncer o no, si aparecieron pólipos colónicos o cáncer de colon durante el seguimiento después de la colectomía, si presentaban adenoma duodenal, pólipos gástricos o tumores desmoides y la existencia o ausencia de enfermedades extracolónicas menos frecuentes (patología de retina, cáncer tiroideo, nódulo tiroideo, adenoma suprarrenal, cáncer de mama), si había antecedentes familiares de PAF y esperanza de vida sin enfermedad.
En el seguimiento de los pacientes, anualmente se solicitaron colonoscopia, gastroduodenoscopia y ecografías de abdomen y tiroides. También se solicitaron de forma sistemática tomografía computarizada (TC) y resonancia magnética (RM) junto con una exploración oftalmológica para confirmar el diagnóstico. En casos sospechosos, estas pruebas se solicitaron con mayor frecuencia y el tratamiento se organizó en consecuencia.
Análisis estadísticoLas variables se presentaron como valores de media ± desviación estándar (DE) o mediana (intervalo, rango intercuartílico [RIQ]) de datos continuos y datos ordinales. Las variables cualitativas se analizaron con la prueba de la χ2 o la prueba exacta de Fisher según correspondiera. Las variables continuas con distribución normal se analizaron con la prueba de la t independiente. La prueba de Kolmogorov-Smirnov se utilizó para evaluar la conformidad de las variables continuas con la distribución normal. La supervivencia se analizó gráficamente con el método de Kaplan-Meyer y las comparaciones se realizaron mediante la prueba del orden logarítmico. Se consideró estadísticamente significativo un valor p bilateral < 0,05. El análisis estadístico se realizó con software disponible comercialmente (IBM-SPSS versión 25.0 para Windows; SPSS).
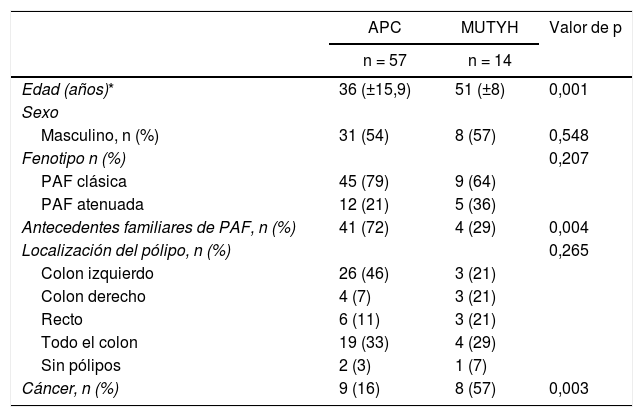
ResultadosDe los 71 pacientes diagnosticados, 14 (20%) presentaban la mutación MUTYH y 57 (80%), la mutación APC (tabla 1).
Datos demográficos y clínicos según las características genéticas de los pacientes con poliposis adenomatosa familiar
| APC | MUTYH | Valor de p | |
|---|---|---|---|
| n = 57 | n = 14 | ||
| Edad (años)* | 36 (±15,9) | 51 (±8) | 0,001 |
| Sexo | |||
| Masculino, n (%) | 31 (54) | 8 (57) | 0,548 |
| Fenotipo n (%) | 0,207 | ||
| PAF clásica | 45 (79) | 9 (64) | |
| PAF atenuada | 12 (21) | 5 (36) | |
| Antecedentes familiares de PAF, n (%) | 41 (72) | 4 (29) | 0,004 |
| Localización del pólipo, n (%) | 0,265 | ||
| Colon izquierdo | 26 (46) | 3 (21) | |
| Colon derecho | 4 (7) | 3 (21) | |
| Recto | 6 (11) | 3 (21) | |
| Todo el colon | 19 (33) | 4 (29) | |
| Sin pólipos | 2 (3) | 1 (7) | |
| Cáncer, n (%) | 9 (16) | 8 (57) | 0,003 |
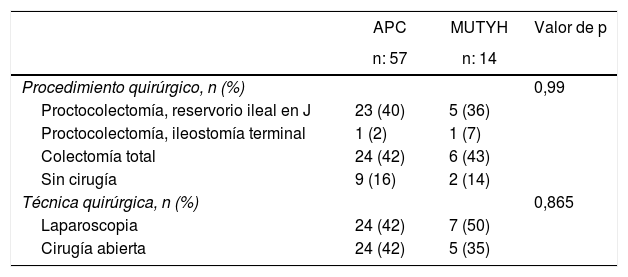
Se solicitó el análisis genético de 11 pacientes (APC: 9 y MUTYH: 2) con antecedentes familiares antes de la aparición de pólipos colónicos y a estos pacientes se les realizó un seguimiento endoscópico. La mutación APC se diagnosticó en pacientes con una media de edad de 22,8 ± 10,8 años. Entre esos casos, a dos de ellos se les diagnosticó afectación extraintestinal (nódulo y cáncer tiroideo). De los 60 pacientes que fueron intervenidos quirúrgicamente, a 30 (50%) se les realizó proctocolectomía y a otros 30 (50%), colectomía total (tabla 2).
Procedimientos quirúrgicos según las características genéticas de los pacientes con poliposis adenomatosa familiar
| APC | MUTYH | Valor de p | |
|---|---|---|---|
| n: 57 | n: 14 | ||
| Procedimiento quirúrgico, n (%) | 0,99 | ||
| Proctocolectomía, reservorio ileal en J | 23 (40) | 5 (36) | |
| Proctocolectomía, ileostomía terminal | 1 (2) | 1 (7) | |
| Colectomía total | 24 (42) | 6 (43) | |
| Sin cirugía | 9 (16) | 2 (14) | |
| Técnica quirúrgica, n (%) | 0,865 | ||
| Laparoscopia | 24 (42) | 7 (50) | |
| Cirugía abierta | 24 (42) | 5 (35) |
APC: Adenomatous polyposis coli; MUTYH: gen mut Y homólogo.
A 14 pacientes (24,5%) se les realizó ampulectomía endoscópica. El procedimiento de Whipple se realizó en dos pacientes (3,5%) con tumores de la ampolla de Vater. En el estudio preoperatorio se diagnósticaron a cuatro pacientes de tumores desmoides. En el resto de los pacientes, los tumores desmoides aparecieron después de la intervención de colon con una media de 93 meses (1-246) y hubo 18 pacientes (58%) con antecedentes familiares previos de tumores desmoides. En 11 pacientes (57,9%) con proctocolectomía y en 12 (60%) con colectomía total apareció un tumor desmoide intraabdominal postoperatorio. Se determinó que no había aparición postoperatoria de tumor desmoide intraabdominal en ocho pacientes (42,1%) con proctocolectomía y en ocho (40%) con colectomía total. No se estableció ninguna diferencia estadísticamente significativa entre estos grupos respecto a la tasa de aparición de tumor desmoide postoperatorio (p = 0,894) (tabla 3).
Enfermedad extracolónica según las características genéticas de los pacientes con poliposis adenomatosa familiar
| APC | MUTYH | Valor de p | |
|---|---|---|---|
| n = 57 | n = 14 | ||
| Adenoma duodenal, n (%) | 36 (63,2) | 3 (21,4) | 0,004 |
| Adenoma duodenal, edad (años)* | 40 (±13,2) | 55 (± 4,1) | |
| Pólipos gástricos, n (%) | 35 (61) | 3(21) | 0,006 |
| Pólipo gástrico, edad (años)* | 41 (± 15,2) | 57 (± 9,4) | |
| Tumor desmoide, n (%) | 31 (54) | 0 | < 0,01 |
| Pared abdominal | 5 (16) | 0 | |
| Intraabdominal | 12 (39) | 0 | |
| Intraabdominal + pared abdominal | 14 (45) | 0 | |
| Tumor desmoide, edad (años)* | 38.9 (± 10.2) | 0 |
Cuando se examinaron otras enfermedades extraintestinales, rara vez se detectaron en pacientes con MUTYH (tabla 4).
Enfermedades extracolónicas según las características genéticas de los pacientes con poliposis adenomatosa familiar
| APC | MUTYH | Valor de p | |
|---|---|---|---|
| n = 57 | n = 14 | ||
| Total de enfermedades extraintestinales, n (%) | 48 (84) | 5 (36) | 0,023 |
| HCEPR, n (%) | 35 (61) | 2 (14) | |
| Cáncer de mama, n (%) | 2 (4) | 0 | |
| Melanoma, n (%) | 2 (4) | 0 | |
| Adenoma suprarrenal, n (%) | 6 (10) | 1 (8) | |
| Nódulo tiroideo, n (%) | 9 (16) | 0 | |
| Cáncer tiroideo, n (%) | 2 (4) | 0 | |
| Cáncer de próstata, n (%) | 2 (4) | 0 | |
| Cáncer de endometrio, n (%) | 2 (2) | 0 | |
| Carcinoma de células basales, n (%) | 0 | 2 (14) |
HCEPR: hipertrofia congénita del epitelio pigmentado de la retina; APC:Adenomatous polyposis coli; MUTYH: gen mut Y homólogo.
En general, se detectó la formación recurrente de pólipos colónicos en 23 pacientes (79,3%) con colectomía total y se realizó una polipectomía endoscópica durante el seguimiento. En 19 pacientes (73%) con mutación APC, a quienes también se les realizó colectomía total, volvieron a aparecer los pólipos colónicos y, por tanto, fue necesaria la polipectomías mediante endoscopias durante el seguimiento. El procedimiento de proctocolectomía se realizó en tres pacientes (13%) porque, durante el seguimiento, se detectaron pólipos en el recto con un número creciente de displasia y pólipos. No se realizaron reoperaciones a pacientes con mutación MUTYH, a quienes con anterioridad se les había realizado colectomía total, y a estos pacientes se les realizó controles endoscópicos (tabla 5).
Resultados a largo plazo en función de las intervenciones quirúrgicas y las características genéticas de los pacientes con poliposis adenomatosa familiar
| APC | MUTYH | Valor de p | |||
|---|---|---|---|---|---|
| n = 57 | n = 14 | ||||
| Intervención quirúrgica | Proctocolectomía | Colectomía total | Proctocolectomía | Colectomía total | |
| n = 24 | n = 24 | n = 6 | n = 6 | ||
| Recidiva de pólipos colónicos, n (%) | 3 (8) | 19 (79) | 1 (17) | 4 (67) | 0,58 |
| Reintervención, n (%) | 0 | 3 (13) | 0 | 0 |
APC: Adenomatous polyposis coli; MUTYH: gen mut Y homólogo.
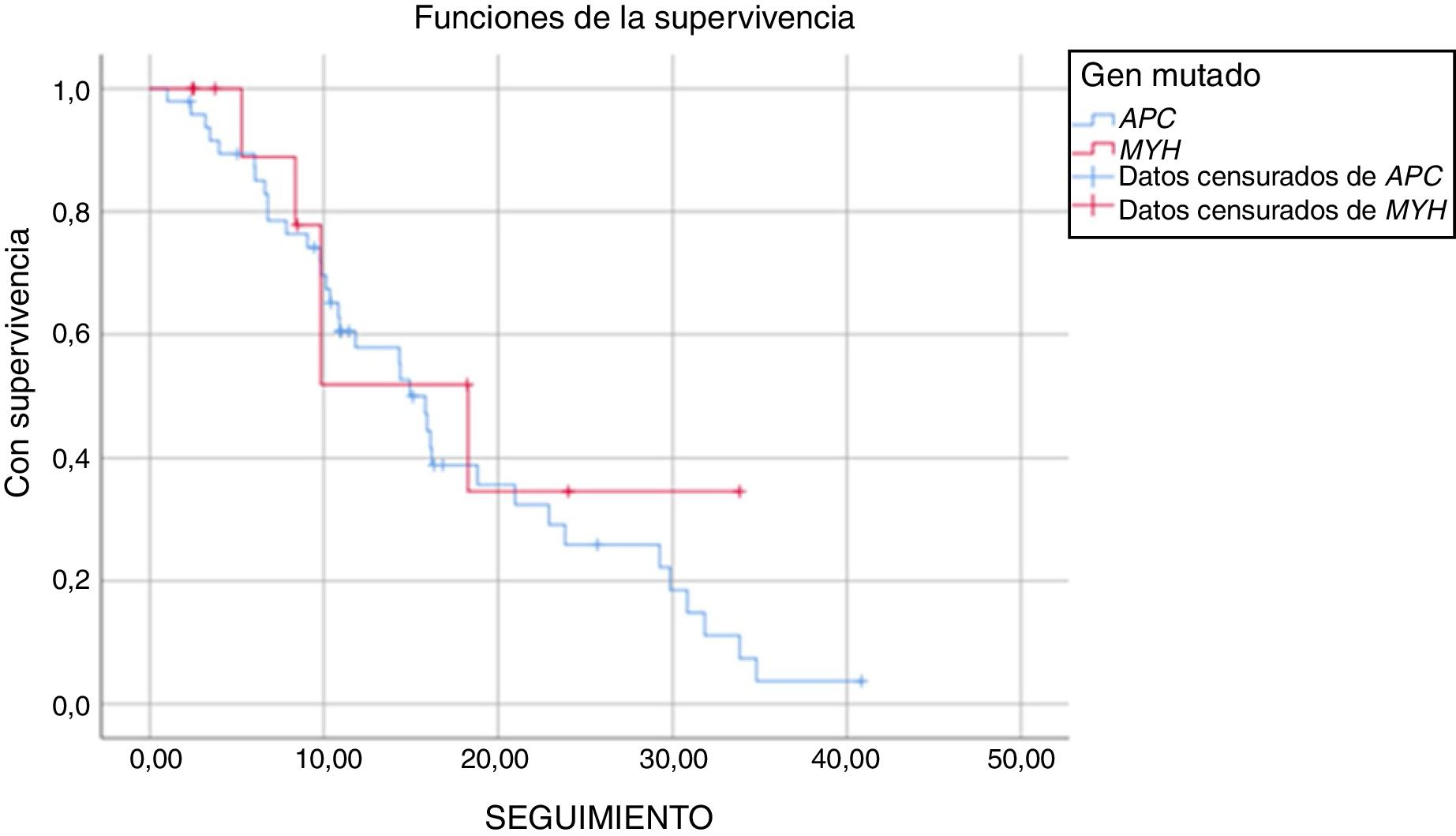
La duración media del seguimiento fue de 17 ± 1,7 años de aquellos pacientes con mutación APC y de 19 ± 4,1 años de aquellos con mutación MUTYH (p = 0,521). Durante el seguimiento de los pacientes con la mutación APC, el 34% no presentaba complicaciones relacionadas con la PAF y el 55% presentaba complicaciones relacionadas con la PAF, que habían surgido a causa de la mutación APC. Durante el período de seguimiento, cinco pacientes con mutación APC fallecieron (un paciente por metástasis de cáncer de colon, un paciente por complicaciones desmoides y tres por otros motivos). De los pacientes con mutación MUTYH, el 57% no presentó enfermedad durante el período de seguimiento y el 43% mostró evolución de la enfermedad durante ese período. En este grupo, se observó mortalidad en cuatro pacientes (tres pacientes por metástasis de cáncer de colon y uno por otros motivos) (fig. 1 y tabla 5).
Discusión
La poliposis colónica es una de las enfermedades más estudiadas desde el punto de vista genético. En general, se han identificado mutaciones del gen APC y del gen MUTYH en pacientes diagnosticados de PAF. El objetivo de este estudio fue contribuir a la mejora del tratamiento de los pacientes con PAF mediante la determinación de la cronología de aparición de las lesiones, el número de pólipos, la afectación extracolónica y el desenlace clínico a largo plazo en los pacientes con mutaciones genéticas.
Los pacientes con mutaciones APC suelen tener más de 100 pólipos en el colon y, en general, presentarán el fenotipo clásico. En la serie actual, el 20% de los pacientes con mutación APC tenían un número de pólipos inferior a 100 y, en contraposición, más pacientes con mutación MUTYH de lo esperado tenían más de 100 pólipos. Contrariamente a lo esperado, no hubo ninguna diferencia estadística entre los dos grupos de pacientes que tenían más de 100 pólipos. La incidencia de más de 100 pólipos de colon en los casos con mutaciones APC4 fue similar a la de estudios anteriores. Por lo general, se comunicó que había menos pacientes con mutación MUTYH con más de 100 pólipos en el colon7,8.
Cuando se analizaron las características anatómicas, se estableció, de acuerdo con la bibliografía, que en pacientes con mutación APC aparecieron pólipos en todo el colon o en el colon izquierdo4. En aquellos con mutación MUTYH, se estableció la misma proporción de pólipos en cada región del colon. Sin embargo, en un estudio se notificó que el colon derecho estaba más afectado cuando se trataba de la mutación MUTYH9. Un número estadísticamente mayor de pacientes con mutación MUTYH se les diagnosticó el tumor en la primera operación en comparación con los pacientes con mutación APC. Los pacientes con mutación APC frecuentemente presentaban antecedentes familares y, por tanto, habían sido sometidos a un análisis genético o exploraciones endoscópicas de forma más precoz, lo que conlleva a un diagnóstico en una fase pre neoplásica de la enfermedad y a una mayor frecuencia de cirugía profiláctica en este grupo de pacientes4.
Cuando se analizaron las características de las enfermedades extracolónicas, se descubrió que el adenoma duodenal y los pólipos gástricos eran estadísticamente bastante más frecuentes en pacientes con mutación APC que en aquellos con mutación MUTYH. Existen diferentes estudios que describen que la tasa de detención de pólipos gástricos era similar en los dos grupos, en cambio la aparición del ampuloma era más frecuentes en el grupo con alteración genética APC10. El ampuloma es una entidad a tener en cuenta en estos pacientes después de la realización de la colectomía profiláctica. La tasa de aparición de mutación APC junto con adenoma duodenal se ha descrito en el 40-90% de los casos11. Al parecer, la media de edad de detección de adenomas duodenales y pólipos gástricos es de 40 años. El diagnóstico de pacientes con mutación MUTYH a una edad más avanzada fue estadísticamente significativo. Inicialmente, las lesiones son benignas pero pueden evolucionar a cáncer con una incidencia de alrededor del 5%12. Dado que el ampuloma inicalmente es una afección benigna, la mejor opción terapéutica para estos pacientes es un método mínimamente invasivo. La ampulectomía endoscópica es la técnica más utilizada en estos pacientes, ya que es la más adecuada si se tiene en cuenta que conlleva una mortalidad y una morbilidad mínimas12,13. Aunque se ha descrito que los adenomas duodenales pueden evolucionar a una neoplasia de la región pancreática en estos pacientes, se ha observado una reducción de la incidencia de estos tumores con la implementación del seguimiento y tratamiento endoscópico14.
El tumor desmoide es la segunda causa más frecuente de muerte en pacientes con poliposis colónica y la mortalidad asociada se presenta, aproximadamente, en el 10%15 de los casos. Los factores de riesgo de los tumores desmoides pueden describirse como mutación de la línea germinal más allá del codón 1444, antecedentes familiares de tumor desmoide y antecedentes personales de cirugía abdominal4,16,17. En estudios anteriores se ha observado que el riesgo de aparición de tumor desmoide no cambia en función del tipo de operación18. En un estudio realizado por Nieuwenhuis et al.19 con 2.260 pacientes, se estableció que no había diferencia respecto a la aparición de tumor desmoide entre los pacientes a quienes se les había realizado colectomía total y aquellos con proctocolectomía. De acuerdo con los hallazgos en la bibliografía, no se estableció una diferencia estadísticamente significativa en el estudio actual respecto a la aparición de tumor desmoide entre los pacientes a quienes se les había realizado colectomía total y aquellos con proctocolectomía. Estudios anteriores15 han comunicado que el riesgo de desarrolar un tumor desmoide durante toda la vida en pacientes con mutación APC es del 10-30%, en cambio en el estudio actual se encontró una incidencia del 54%. Esto puede atribuirse a los pacientes del estudio actual, con una tasa del 58% de antecedentes familiares de tumor desmoide, y también podría estar asociado con las ubicaciones de la mutación genética. La media de edad de aparición del tumor desmoide se estableció en 39 años. En el estudio actual, la tasa de incidencia del diagnóstico del tumor desmoide antes de realizar una colectomía profiláctica fue del 13% y se constató que este resultado era relativamente más alto que las tasas que aparecían en estudios anteriores4,16. En pacientes con la mutación APC se diagnosticó una tasa estadísticamente bastante mayor de enfermedades extracolónicas17.
El carcinoma tiroideo asociado con PAF es el tercer tipo de cáncer más frecuente, con un riesgo de desarrollo durante la vida del 1-2%20. En los exámenes anuales realizados en el estudio actual, se determinó una tasa del 4% de carcinoma tiroideo y del 16% de nódulos tiroideos. El adenoma suprarrenal puede aparecer en una tasa del 7-13% de los pacientes con PAF20 y, de acuerdo con estas cifras en la bibliografía, apareció adenoma suprarrenal en el 10% de los pacientes de la serie actual. La hipertrofia congénita del epitelio pigmentado de la retina (HCEPR) es una lesión senil que se caracteriza por la aparición de pigmentación en el fondo del ojo. Es un marcador fenotípico de PAF con una prevalencia observada del 70-75%21,22. En la serie actual, se estableció en el 61%, una tasa menor que los hallazgos anteriores en la bibliografía.
En el tratamiento de pacientes con colectomía profiláctica, se ha observado que el cáncer aparece alrededor de los 39 años. Por tanto, en estos paciente la mejor opción terapéutica es la cirugía profiláctica precoz para garantizar un buen desenlace clínico futuro14. El objetivo de la colectomía profiláctica es mantener la mejor calidad de vida posible y proteger al paciente del cáncer colorrectal23,24. En los últimos años, en nuestros pacientes se han preferido la proctocolectomía laparoscópica, la mucosectomía y el reservorio ileal en J. En la proctocolectomía, la calidad de vida es mucho más baja que la que se obtiene con la colectomía total25. La colectomía total se puede realizar si se ha verificado la mutación y la colonoscopia preoperatoria no establece que la localizacion de los pólipos sea predominante en el recto. La proctocolectomía se realizó antes de que se estableciera la mutación genética debido a la existencia de múltiples pólipos de histología dudosa, que no podían controlarse mediante endoscopia, y también debido al riesgo de que los múltiples pólipos displásicos degenerasen en un cáncer.
A pesar del mayor número de reoperaciones en pacientes con colectomía total que en aquellos con proctocolectomía, no se encontraron diferencias estadísticamente significativas entre ambos. En la población de estudio de pacientes con poliposis de un solo centro, incluso con una definición de mutación más amplia, sólo encontramos que en el 23,9% de los pacientes se sospechaba mutación PAFA. Este bajo número de pacientes dificulta hallar una definición precisa de este síndrome y establecer las pautas de tratamiento. En los pacientes en quienes se sospecha PAFA, se recomienda la conservación del recto ya que se cree que los pólipos rectales se producen con menos frecuencia4,21. Se debe tener la precaución de realizar un seguimiento riguroso de los pacientes y, si se observan pólipos en el recto, se puede realizar una proctocolectomía como segundo procedimiento quirúrgico.
Entre las limitaciones de este estudio se encuentran el diseño retrospectivo y el hecho de que no se especificaron las características de los pacientes en función de la localización de la mutación.
En conclusión, las pruebas genéticas de pacientes con PAF serán útiles para establecer el desenlace clínico futuro y servirán de guía para decidir el tipo de tratamiento quirúrgico.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.

