




El síndrome de Sagliker (SS) fue descrito por primera vez en 20041. Esta entidad única e infrecuente, se presenta en pacientes con enfermedad renal crónica (ERC) en el curso de un hiperparatiroidismo secundario (HPTS) inadecuadamente tratado2. Se estima que la incidencia del SS es del 0,5% de los pacientes en hemodiálisis1. El SS es una manifestación exagerada de osteodistrofia renal que combina la ERC avanzada, el HPTS no controlado y una deformación de los rasgos faciales junto con la aparición de tumores pardos3. Tras el trasplante renal, la deformidad facial no es reversible y ello conlleva una disminución de la calidad de vida de los pacientes afectados4–6.
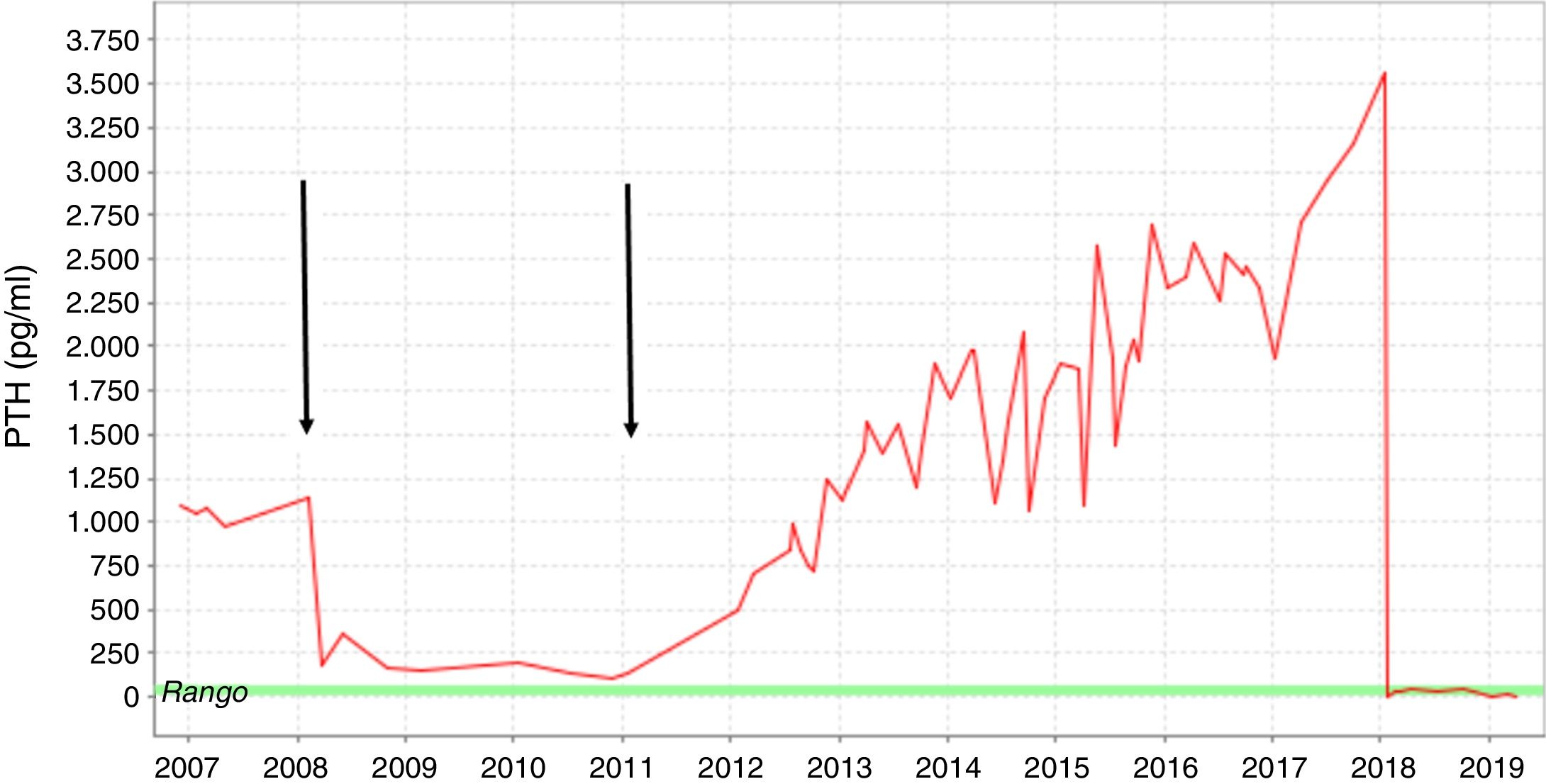
Presentamos el caso de un varón de 35 años con ERC secundaria a glomeruloesclerosis en diálisis durante 9 años, que refería cambios inusuales en sus rasgos faciales en los últimos años. La pérdida completa de la función renal residual ocurrió en 2007. Un año antes, se detectó un aumento progresivo de los niveles de parathormona (PTH). El paciente recibió calcimiméticos, quelantes de fosfato y vitamina D, sin embargo, los niveles de PTH continuaron elevándose. En 2008, el paciente fue sometido a un trasplante renal de donante cadáver. Durante este periodo, los valores de PTH descendieron hasta alcanzar niveles casi normales. En 2010, se observó un descenso de la función renal del injerto debido a una baja adherencia al tratamiento inmunosupresor. Al principio, el paciente rechazó entrar en el programa de hemodiálisis. Finalmente, en 2012 lo reinició. En ese momento, los niveles de PTH se habían elevado hasta 1.000pg/ml (fig. 1).
 en el curso de la diálisis.")
Fluctuación del nivel de la PTH desde 2007 hasta 2019. Las flechas indican el trasplante renal realizado en 2008 y el rechazo renal agudo sufrido por el paciente antes de reanudar el programa de hemodiálisis. Se usó medicación farmacológica para el HPTS (calcimiméticos, aglutinantes de fosfato y vitamina D) en el curso de la diálisis.
Durante los 6 años siguientes, los niveles de PTH continuaron elevándose desde 1.000 hasta 3.500pg/ml a pesar de un tratamiento intensivo que incluía sevelamer 3.200mg/8h, paricalcitol 9μg/IV/3 veces en semana y cinacalcet 90mg/8h.
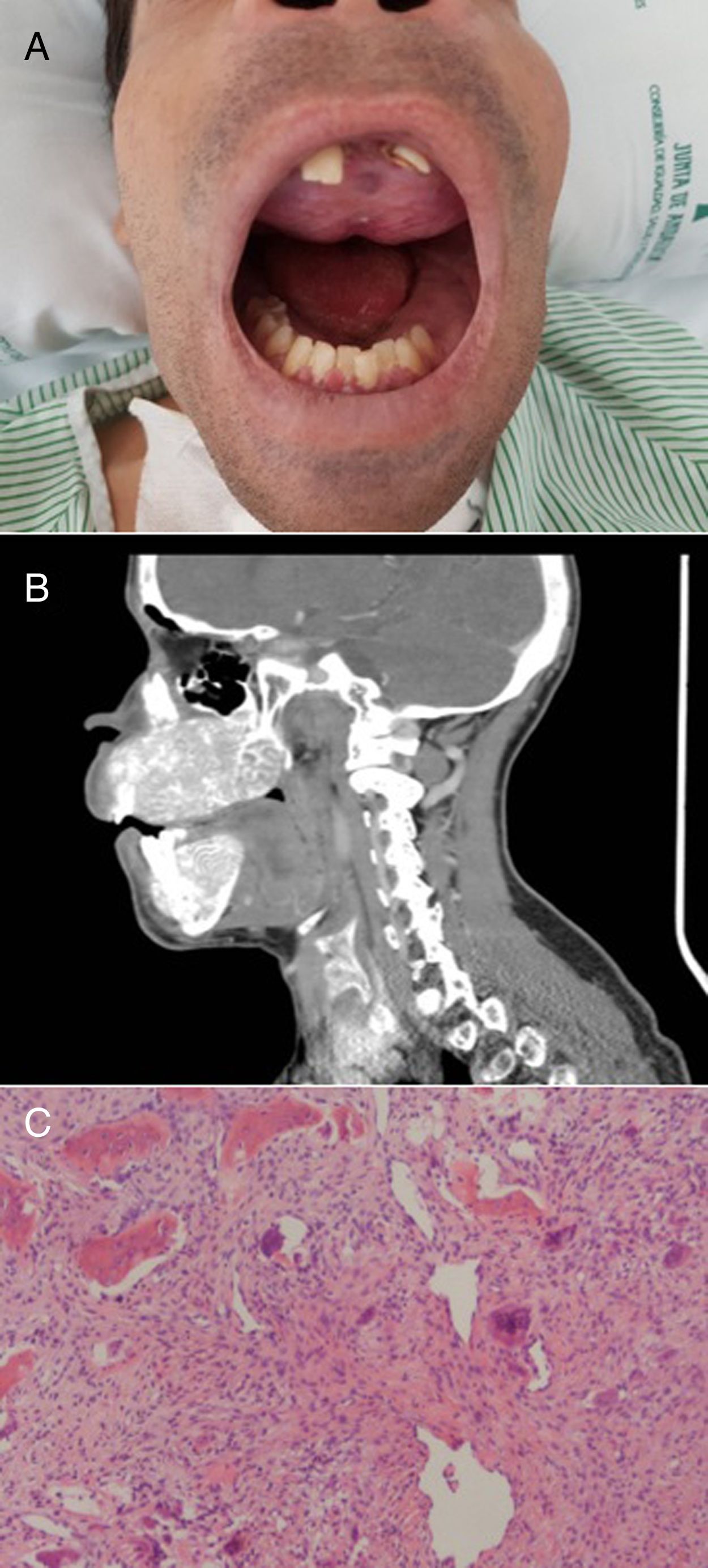
El paciente refería dolor de cabeza, fatiga, dolores articulares y cambios en la punta de los dedos. Su rostro mostraba una marcada protuberancia frontal y deformidades obvias tanto del maxilar como de la mandíbula. Ambos huesos se expandieron notablemente con una pérdida de la arquitectura normal, haciendo que sus dientes se movieran y presentaran una disposición irregular (fig. 2A). La tomografía de cráneo mostró afectación difusa de la base y la bóveda craneal (fig. 2B). La ecografía cervical reveló 2 lesiones nodulares extratiroideas debajo del polo inferior del lóbulo tiroideo derecho y del polo inferior del lóbulo tiroideo izquierdo. En el SPECT/TAC se observó como único hallazgo una retención patológica del radiofármaco sobre el extremo inferior del lóbulo izquierdo del tiroides, sugestiva de adenoma de paratiroides.
 Deformidad del maxilar y la mandíbula. Tumor pardo en el paladar superior. B) TAC del cráneo que muestra una enfermedad ósea difusa caracterizada por la deformidad obvia de la mandíbula superior e inferior, desmineralización del esqueleto axial y lesiones extensas de apariencia lítica. C) Células gigantes mesenquimales y multinucleadas, típicas del tumor pardo (H&E, ×50).")
A) Deformidad del maxilar y la mandíbula. Tumor pardo en el paladar superior. B) TAC del cráneo que muestra una enfermedad ósea difusa caracterizada por la deformidad obvia de la mandíbula superior e inferior, desmineralización del esqueleto axial y lesiones extensas de apariencia lítica. C) Células gigantes mesenquimales y multinucleadas, típicas del tumor pardo (H&E, ×50).
En 2017, tras la biopsia del paladar superior se confirmó la presencia de un tumor pardo (fig. 2C). Las deformidades características del rostro junto con la aparición de un tumor pardo confirmaron el diagnóstico de SS.
El paciente fue sometido a una paratiroidectomía total con autoimplante de un fragmento de una de las glándulas y timectomía. Durante el postoperatorio, el paciente presentó una hipocalcemia de difícil control con necesidad de calcitriol, calcio carbonato oral y gluconato cálcico intravenoso durante 5 días. Tras un año de seguimiento, su nivel de calcio permanecía estable y su nivel de PTH fue de 14pg/ml. El paciente refería sentirse subjetivamente mejor y una leve mejoría de sus rasgos faciales, aunque no era capaz de realizar la oclusión de su boca.
Los mecanismos por los cuales algunos pacientes con ERC desarrollan SS son desconocidos. Algunos autores sugieren que puede deberse a una alteración genética que se desencadena durante la diálisis. Un estudio internacional sugirió que las mutaciones del gen GNAS1 podrían promover la génesis del SS4. El HPTS evolucionado y el tratamiento inapropiado pueden desempeñar un papel importante en la aparición del SS3,4. En nuestro paciente, la falta de adherencia al tratamiento que llevó a un rechazo del injerto, y su negativa a someterse a hemodiálisis pudieron ser los detonantes de su enfermedad.
Los cambios faciales en el hiperparatiroidismo se asocian exclusivamente con los pacientes con ERC avanzada y su grado de asociación depende en gran medida de la severidad de la enfermedad y la duración de la misma1. Nuestro paciente desarrolló deformidades en su cráneo y mandíbula a pesar de estar expuesto a dosis máximas de cinacalcet. Cinacalcet es un fármaco extremadamente caro, y en aquellos casos con glándulas paratiroides con hiperplasia nodular y un volumen >500mm3 parece asociarse a una resistencia al tratamiento3, como ocurrió con nuestro paciente.
Según las guías clínicas Kidney Disease Improving Global Outcomes (KDIGO) aquellos pacientes que presentan desde un moderado deterioro de la función renal (G3a) hasta un fallo renal (G5d), con un importante hiperparatiroidismo secundario que no responde al tratamiento médico, deben someterse a una paratiroidectomía7. La postergación de la paratiroidectomía durante años claramente contribuyó a la aparición de las alteraciones funcionales y cosméticas de su rostro y sus manos, y a la presencia de un síndrome de hueso hambriento de difícil control con una estancia prolongada en el postoperatorio.
Por ello, en aquellos pacientes con ERC e HPTS inadecuadamente tratado2, la paratiroidectomía debe realizarse sin demora, antes de la aparición o indicios del SS.

