




Ehlers–Danlos syndrome (EDS) is a group of rare hereditary disorders (1:5000 newborns) that are characterized by cutaneous hyperextensibility, joint hypermobility and connective tissue fragility. Within the forms of presentation, aneurysmal rupture is rare; while it is a characteristic feature in the vascular variation, it is exceptional in other types.1 We present the case of a 38-year-old woman diagnosed with classical EDS as a result of hemoperitoneum secondary to the rupture of a giant aneurysm of the splenic artery.
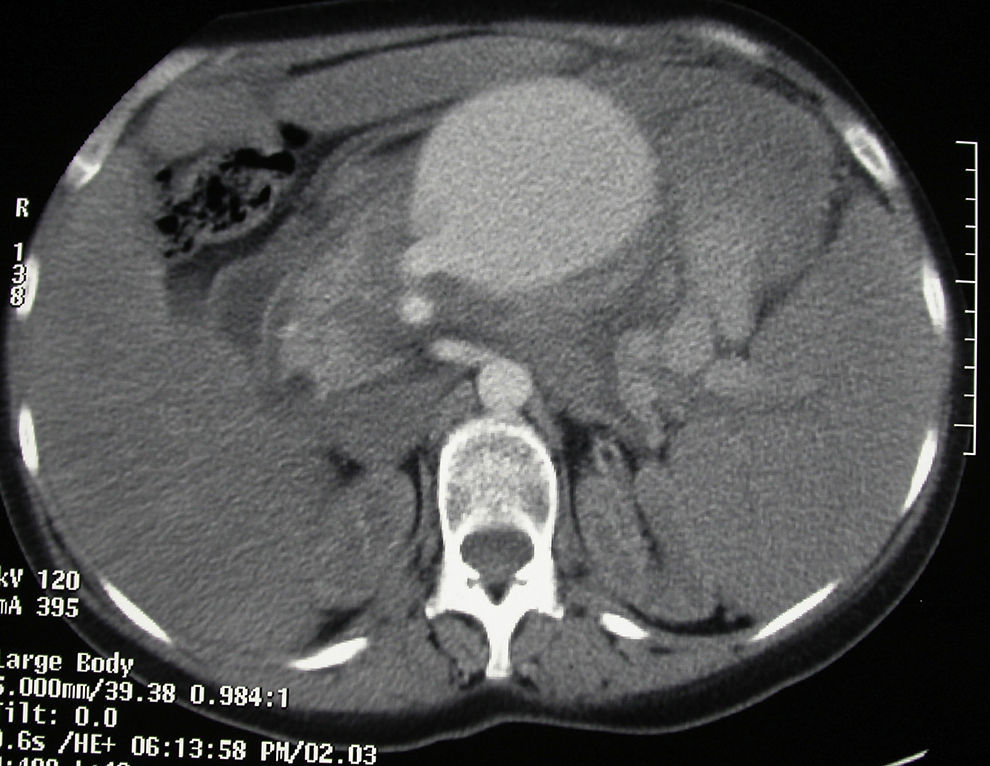
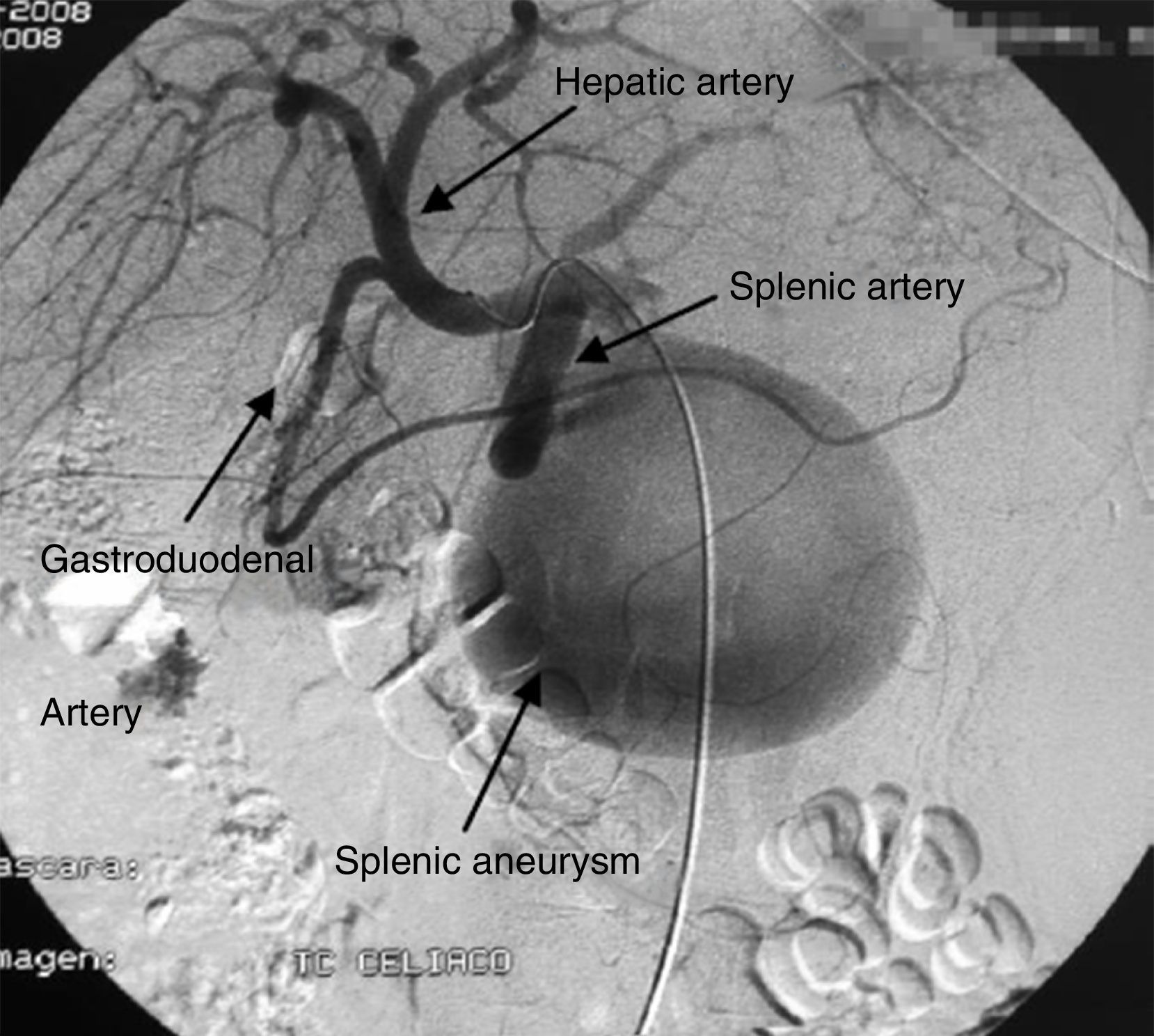
The patient came to the emergency room with abdominal pain and vomiting that was not associated with any previous trauma. Her medical history included renal failure and autoimmune hypothyroidism. Upon arrival to the ER, the patient was hemodynamically unstable. Physical examination detected a distended abdomen that was painful to palpation, associated with splenomegaly. Lab work showed hematocrit 10% and hemoglobin 3.3g/dl. Abdominal CT scan (Fig. 1) and angiography (Fig. 2) revealed a large aneurysm 4cm from the origin of the splenic artery and 2 pseudoaneurysms located in the splenic hilum. The largest was embolized with thrombin and metallic coils. In spite of the interventionist treatment, the patient remained hemodynamically unstable, so we decided to operate. We discovered a hemoperitoneum of more than 2l with splenomegaly (approximately 30cm) and a pseudoaneurysm over the body of the pancreas that was partially ruptured at the inferior pole of approximately 10cm. Hilar dissection was conducted with transfixing ligatures and a standard splenectomy was performed. The patient progressed favorably and was discharged on the tenth day post-op.


The patient presented leptosomic appearance and a marfanoid phenotype, so she was referred to the medical genetics department. Genetic studies detected a mutation in the COL5A1 gene (c.1588G>A) that, at the protein level, changes the glycine in position 530 for serine (p.Gly530ser).
According to Villefranche, EDS is classified into 6 main subtypes. The classical type, the type associated with hypermobility and the vascular type are the most common, while kyphoscoliosis, arthrochalasis and the dermatosparaxis types are less common conditions. Most EDS forms correspond with mutations in genes coding for collagen chains or enzymes involved in the biosynthesis of these proteins.1 In the classic type (around 50% of cases), the defect is caused by mutations in the genes coding for V alpha-1 (COL5A1) or alpha-2 (COL5A2) collagen.2,3 When EDS is suspected, we should review the patients’ medical history for complications, such as frequent hematomas, cervical insufficiency, anal prolapse in childhood, Premature rupture of membranes, vaginal lacerations, low lung capacity or murmurs.3,4 The frequency and severity of the digestive symptoms in EDS depends on the type, and it is the vascular type that presents greater gastrointestinal complications. There are few reports in the literature about cases of spontaneous splenic, hepatic or biliary rupture, probably due to a smaller quantity of structurally intact support tissue in these organs.5
In the differential diagnosis of acute abdominal pain in EDS, the vascular system should always be considered. The majority of EDS subtypes have bleeding diathesis, in spite of normal coagulation profiles. The forms of presentation can be mild, such as hematomas, or more severe, such as arterial dissections, aneurysms and spontaneous ruptures. The most severe from is seen in the vascular type,6 where the complications are mainly arterial, involving intermediate-sized abdominal vessels (renal, iliac, femoral, mesenteric and hepatic)7 and the abdominal aorta, although they have also been described in the classical type. Invasive diagnostic methods should be avoided as they could cause serious complications due to the fragility of the vessels.4 It is recommended to use non-invasive methods, such as ultrasound, CT angiogram or MRI. Unlike in our case, angiography should be avoided as it is associated with a rate of complications between 17 and 67% and a mortality rate of 6%–19%.8
In the surgical treatment of vascular disorders in patients with EDS, currently both open and endovascular procedures are recognized as having good results,1 but few patients have undergone emergency endovascular treatments.2 In case of hemorrhage, it is recommended to be as conservative as possible by administering desmopressin, compression and the use of local hemostatic measures and adhesives.9 The following step involves the use of endovascular methods, such as embolization or stent/prosthesis repair. As in our case, when less invasive measures fail, surgery is necessary.4
To date, the 5 cases described of spontaneous rupture of the splenic artery secondary to aneurysm are associated with EDS of the vascular type, with a mortality rate of 25%.10 The case that we present is the first of rupture of the splenic artery associated with classic-type EDS reported in the literature.
With this case, we have tried to emphasize the importance of overall patient assessment, where both the patient medical history as well as the physical examination enables us to correlate intraoperative findings with the final diagnosis.
Please cite this article as: Paredes Quiles M, López V, Fernández JÁ, Cascales P, Sánchez Valero J, Parrilla P. Rotura espontánea de aneurisma de arteria esplénica en el síndrome de Ehlers–Danlos tipo clásico. Cir Esp. 2016;94:543–544.
