




Thyroid hormones have a significant impact on heart function, mediated by genomic and non-genomic effects. Consequently, thyroid hormone deficiencies, as well as excesses, are expected to result in profound changes in cardiac function regulation and cardiovascular hemodynamics. Thyroid hormones upregulate the expression of the sarcoplasmic reticulum calcium-activated ATPase and downregulate the expression of phospholamban. Overall, hyperthyroidism is characterized by an increase in resting heart rate, blood volume, stroke volume, myocardial contractility, and ejection fraction. The development of “high-output heart failure” in hyperthyroidism may be due to “tachycardia-mediated cardiomyopathy”. On the other hand, in a hypothyroid state, thyroid hormone deficiency results in lower heart rate and weakening of myocardial contraction and relaxation, with prolonged systolic and early diastolic times. Cardiac preload is decreased due to impaired diastolic function. Cardiac afterload is increased, and chronotropic and inotropic functions are reduced. Subclinical thyroid dysfunction is relatively common in patients over 65 years of age. In general, subclinical hypothyroidism increases the risk of coronary heart disease (CHD) mortality and CHD events, but not of total mortality. The risk of CHD mortality and atrial fibrillation (but not other outcomes) in subclinical hyperthyroidism is higher among patients with very low levels of thyrotropin. Finally, medications such as amiodarone may induce hypothyroidism (mediated by the Wolff–Chaikoff), as well as hyperthyroidism (mediated by the Jod–Basedow effect). In both instances, the underlying cause is the high concentration of iodine in this medication.
Las Hormonas Tiroideas (HT) tienen un impacto significativo sobre la función cardiaca, el cual es mediado por efectos genómicos y no-genómicos. Como consecuencia, la deficiencia y el exceso de las HT origina profundos cambios en la regulación de la función cardiaca y en algunos aspectos hemodinámicos y cardiovasculares. Las HT supra-regulan la expresión de la ATPasa activada por calcio del retículo sarcoplasmático, e infra-regulan la expresión de fosfolambán. En general, el hipertiroidismo se caracteriza por un incremento en la frecuencia cardiaca en reposo, del volumen sanguíneo, de la contractilidad miocárdica y del volumen sistólico, entre otros. El desarrollo de “Falla cardiaca de alto gasto” en hipertiroidismo puede ser debido a “Cardiomiopatía mediada por taquicardia”. Por otro lado; en el estado hipotiroideo, la deficiencia de HT origina bradicardia, debilidad en la contractilidad y relajación miocárdica, con prolongación del tiempo sistólico y diastólico temprano. La disminución en la precarga se debe a las alteraciones en la función diastólica; la post-carga se incrementa, y las funciones cronotrópicas e inotrópicas están disminuidas. El hipotiroidismo subclínico incrementa el riesgo de mortalidad por Enfermedad Arterial Coronaria (EAC) y de eventos por EAC, pero no aumenta el riesgo de mortalidad total. El riesgo de mortalidad por EAC y de fibrilación auricular (pero no de otros resultados) en hipertiroidismo subclínico es mayor entre pacientes con niveles muy bajos de tirotropina. Finalmente, medicamentos como la amiodarona puede inducir hipotiroidismo (mediado por el efecto de Wolff-Chaikoff, además de hipertiroidismo (mediado por el efecto de Jod-Basedow. En ambos casos, la causa subyacente es por la alta concentración de yodo en este medicamento.
It is likely that all cells in the body are targets for thyroid hormones (TH). Despite not being strictly necessary for life, TH have profound effects on many physiologic processes. Changes in thyroid status markedly influence cardiac contractile and electrical activity; increased or reduced action of TH on certain molecular pathways in the heart and vasculature causes relevant cardiovascular derangements. Receptors for TH are intracellular DNA-binding proteins that function as hormone-responsive transcription factors; TH enter cells through membrane transporter proteins. A number of plasma membrane transporters have been identified, some of which require ATP hydrolysis; once inside the nucleus, the hormone binds to its receptor, and the hormone-receptor complex interacts with specific DNA sequences in the promoter regions of responsive genes. The effect of DNA-binding of the hormone-receptor complex is to modulate gene expression, either by stimulating or inhibiting transcription of specific genes. Cellular actions of TH may be initiated within the cell nucleus, at the plasma membrane, in cytoplasm and cytoskeleton, and in organelles; changes in gene expression caused by TH have a significant effect on the contractile apparatus and the sarcoplasmic reticulum. Consequently, it is expected that TH excess or deficit will be reflected in increased myocardial contractility, heart rate, relaxation, arrhythmias and cardiac output (in hyperthyroidism), and decreases in these parameters in hypothyroidism.
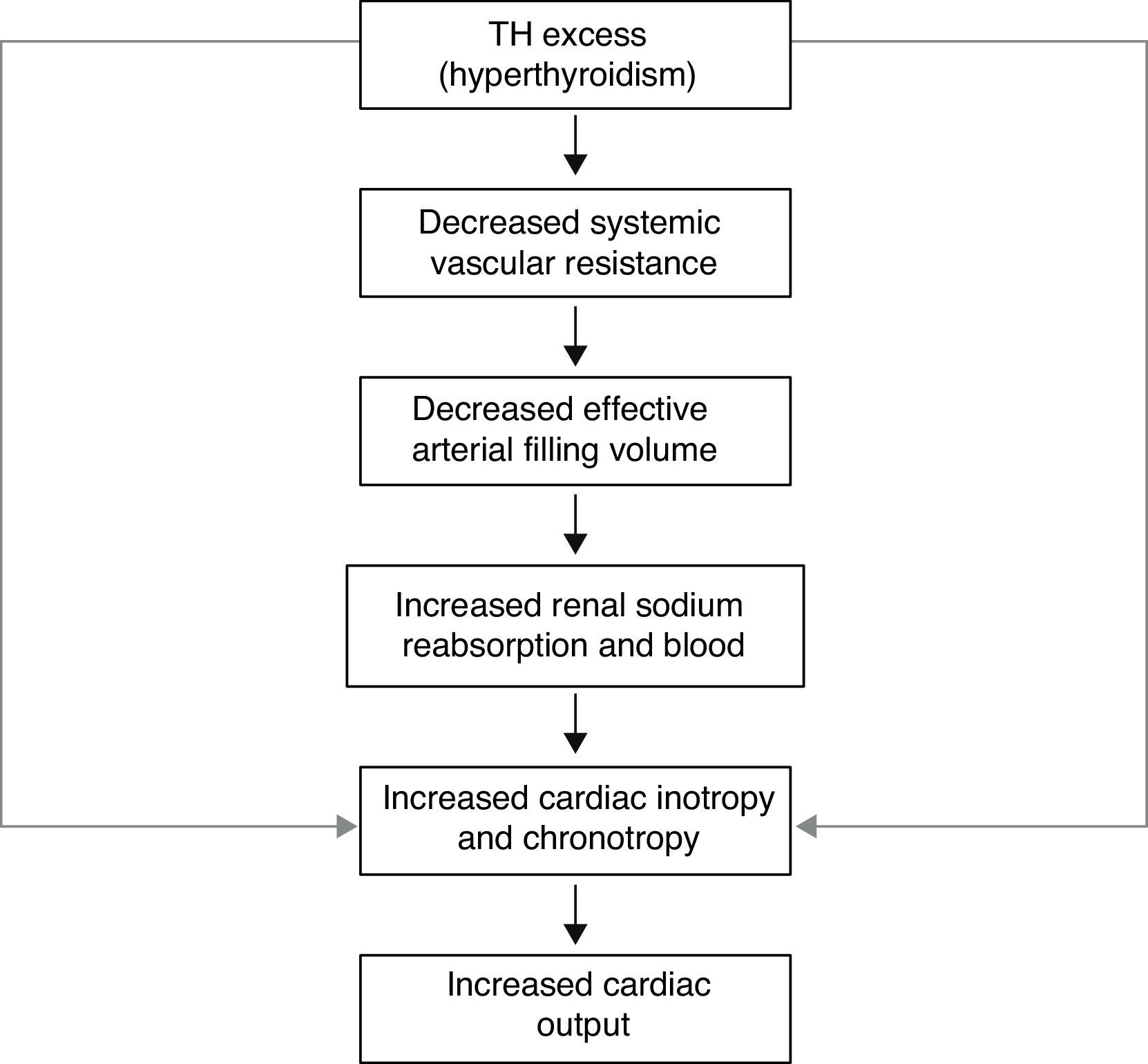
Thyroid hormone nuclear receptors (TRs) mediate the biological activities of T3 via transcriptional regulation, and the genes that are transcriptionally regulated by T3 are critical in the regulation of systolic and diastolic properties of the myocardium. T3 is the biologically active thyroid hormone; it is mostly generated peripherally by 5′-monodeiodination of thyroxine (T4). TH have a pro-angiogenic effect in adults and can stimulate arteriolar growth in the normal heart as well as after myocardial infarction. In presence of hyperthyroidism, the preload is increased; there is high cardiac output, with increased heart rate, reduced peripheral vascular resistance and hyperdynamic circulation. The reduction in systemic vascular resistance is responsible for the decrease in renal perfusion pressure and for activation of the Renin–Angiotensin–Aldosterone system (RAAS), with the resulting increase in sodium absorption and blood volume. The increased risk of cardiac mortality could be a consequence of the increased risk of arrhythmias, especially atrial fibrillation (AF), and the risk of heart failure in these subjects. In presence of hypothyroidism, there are important changes in cardiac structure and function, with severity depending on the degree and the duration of TH deficiency. This state is characterized by low cardiac output, decreased heart rate and stroke volume, reduction in systolic and diastolic functions; there is also a decline in cardiac preload and blood volume, as well as a drop in renal perfusion with impaired free water clearance and hyponatremia. The low cardiac output is caused by bradycardia and a reduction in ventricular filling and cardiac contractility. Systemic vascular resistance may increase, and diastolic relaxation and filling are slow. An increase in cardiovascular risk and mortality has also been described.
Mechanisms of TH action on the cardiovascular systemTH play a key role in energy homeostasis. The set point for TH production and secretion by the thyroid gland is regulated by the hypothalamic thyrotropin-releasing hormone (TRH), determining the equilibrium between serum thyroid stimulating hormone (TSH) and TH concentrations.1,2 The major form of TH produced by the thyroid is the pro-hormone thyroxine (T4) which can be converted into the biologically active tri-iodothyronine (T3) mediated by the removal of an iodide by deiodinases (the iodothyronine deiodinases constitute a family of selenoenzymes that selectively remove iodide from thyroxine and its derivatives, thus activating or inactivating these hormones which have a tissue-specific distribution). All deiodinases are membrane-anchored proteins of 29–33kDa that share substantial sequence homology, catalytic properties and contain selenocysteine as the key residue within their catalytic centers; they catalyze and sequentially remove stereo-specific iodine atoms from T4, generating active and inactive isomers of both T3 and diiodothyronine (T2). The deiodination of T4, T3, and other iodothyronines is an integral component of TH homeostasis.3,4 There are three deiodinases: Type 1 (D1), localized to the plasma membrane and expressed in liver, thyroid and kidney, it catalyzes removal of inner or outer ring iodine atoms in equimolar proportions to generate T3, reverse T3 (rT3), or T2, depending on the substrate. Most of the circulating T3 is derived from conversion of T4 to T3 by the actions of D1. Type 2 (D2), which is considerably more efficient than D1, catalyzes only the removal of an outer ring iodine atom from T4, generating the active product T3. The major role of D2 is to control the intracellular T3 concentration, its availability to the nucleus, and the saturation of the nuclear T3 receptor in target tissues; it is mainly active in brain, pituitary, and skeletal muscle. And Type 3 (D3), which is expressed in the brain and other tissues; it irreversibly inactivates T3, or prevents activation of T4 by catalyzing removal of an inner ring iodine atom to generate T2 or rT3, respectively. Thus, inactivating D3 prevents TH access to specific tissues at critical times and reduces TH receptors (TRs) saturation. Given these functions, D3 is considered the major physiological inactivator and terminator of TH action at the peripheral level.5,6
TH signaling is a local phenomenon, with target cells playing a major role through restricted expression of the activating or inactivating deiodinases. This local role played by the deiodinases in customizing TH signaling is the main way in which TH exert its metabolic effects. Although the thyroid gland produces predominantly T4, the primary biologically active form of the hormone is T3, which binds to, and activates, the TRs. TRs homodimerize or interact with other nuclear receptors such as the retinoic X receptor and control essential functions in growth, development and metabolism, and are important for normal functioning of almost all tissues.
TRs are transcription factors that bind to thyroid hormone response elements (TREs) in the regulatory regions of target genes. TRs are encoded by two genes, THRA and THRB, located on chromosomes 17 and 3, respectively.
T3, the active form of TH, exerts many of its actions through its TRs: TRα1, TRα2, TRβ1, and TRβ2. TRs, with the exception of TRβ2, are expressed in all tissues and the pattern of expression varies in different types of tissues. TRα1 is predominantly expressed in the myocardium and regulates important genes related to cell differentiation and growth, contractile function, pacemaker activity, and conduction. The three major TRs isoforms, TRα1, TRβ1, and TRβ2, are expressed in a tissue-specific fashion and regulate a spectrum of metabolic and developmental functions; moreover, TRs are members of the nuclear receptor superfamily and function as T3-inducible transcription factors.7,8
The TRs are highly homologous transcription factors which transduce signals of active forms of TH (especially T3). Like other nuclear receptors, TRs bind TREs comprised of degenerate repeats of the sequence AGGTCA, usually as heterodimers with retinoid X receptors (RXRs). From these locations, the TRs recruit co-regulator complexes that influence gene expression, and T3 modulates transcription by inducing conformational changes in the receptor C-terminal ligand binding domain which, in turn, alters the complement of TR associated co-regulators.
The selective actions of TRs are influenced by local ligand availability, by transport of TH into the cell by related transporters, by the relative expression and distribution of the TR isoforms and nuclear receptor co-repressors and co-activators; and, finally, by the sequence, arrangement, and promoter context of the TREs.
TH binds to serum transport proteins that help ensure even delivery of hormone to all tissues, cell type-specific membrane transporters, cytoplasmic interacting proteins, enzymes that variously activate pro-hormones or inactivate active hormones, and the TRs themselves. TH interacting proteins regulate important steps that dictate hormone availability for the intracellular receptor, so it is very important to understand structure activity relationships of TH in the context of both the receptor and the proteins that comprise the entire signaling system.
TH acts in large part by binding to nuclear TRs. Binding of the T3 ligand to the TRs results, for a great majority of genes, in their increased transcription. In the absence of the T3 ligand, TRs can repress the expression of genes leading to gene silencing. The communication between the TH and the basal transcription machinery occurs through a complex set of co-activators and co-repressors.9,10
The ligand-activated TRs recruit co-activators, which have a positive stimulatory interaction with the basic transcriptional machinery. In contrast, recruitment of TH-related co-repressors leads to decreased transcription of TH-responsive genes.
The mediation of nuclear T3 receptor-based TH action is a complex process which is influenced by TH concentration and the level and type of the TR alpha and beta isoforms; the configuration of the TREs also influences TH action.
The important role of interactions with co-repressors and co-activators, and the interactions with cell type-specific factors, lead to changes in the histone acetylation status of chromatin.
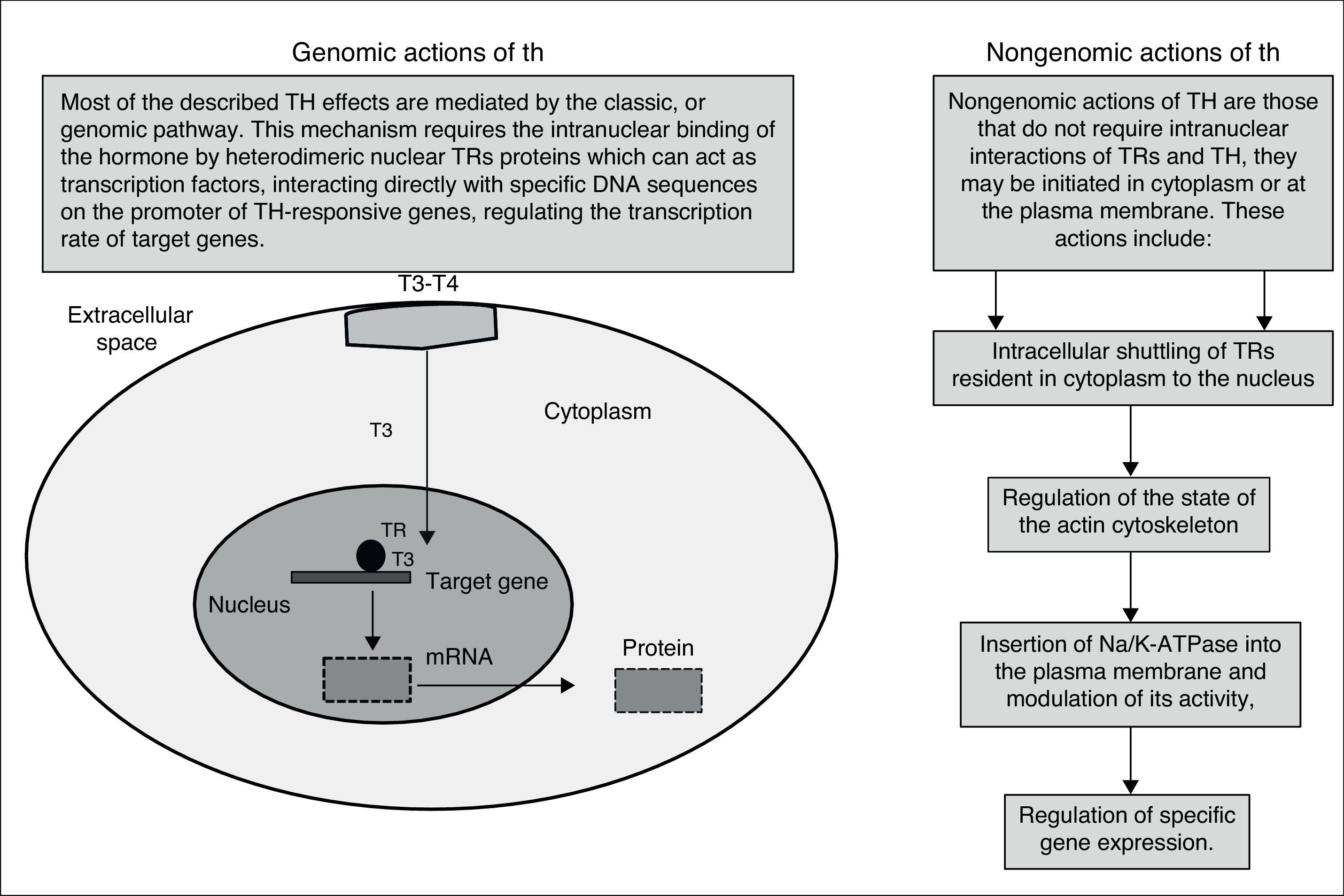
Besides the well-characterized genomic action (nuclear) of TH, mediated by TRs, the non-genomic action of TH is often related to activation of signaling pathways. The classical genomic actions of T3 are mediated by high-affinity nuclear receptors that regulate gene expression directly. This process begins with the entry of T3 into the cardiomyocyte through specific transport proteins located within the cell membrane; once in the cardiomyocyte, T3 enters the nucleus and interacts with specific transcriptional co-activators or with co-repressors. Occupancy of these receptors by T3, in combination with recruited co-factors, allows the TR complex to bind or release specific DNA sequences (TREs).
In contrast, the non-genomic effects of TH occur rapidly and are unaffected by transcription inhibitors and protein synthesis. The genomic actions of TH have an established role in the development, differentiation and homeostatic maintenance of target tissues.11,12
T3 exerts its effects by two mechanisms: genomic actions consisting of T3 link to nuclear receptors that bind responsive elements in the promoter of target genes, and the extranuclear nongenomic activities on the cardiac myocyte and on the systemic vasculature can occur rapidly and do not involve TREs-mediated transcriptional events (Fig. 1). These activities include rapid effects on the plasma membrane and cytoplasmic organelles. Many of the rapid effects mediated by these hormones are not changed by the use of transcription and translation inhibitors; however, these T3-mediated effects include changes in various membrane sodium, potassium, and calcium ion channels, effects on actin polymerization, and on the intracellular signaling pathways in the heart and vascular smooth muscle cells. Both the non-genomic and genomic effects of T3 act in concert to regulate cardiac function and cardiovascular hemodynamics.

Moreover, myocardial contraction and relaxation are mediated through the release and re-uptake of calcium, respectively. Some abnormalities of cardiac function in patients with thyroid dysfunction directly reflect the effects of TH on calcium-activated ATPase and phospholamban, which are involved primarily in the regulation of systodiastolic calcium concentrations in cardiomyocytes. Calcium re-uptake is dependent on the action of sarcoplasmic Ca2+-ATPase, which is normally inhibited by phospholamban. Phosphorylation of phospholamban inhibits the action of phospholamban and results in increased affinity of the Ca2+-ATPase for calcium, increased calcium uptake, and diastolic relaxation. Sarcoplasmic reticulum calcium-activated ATPase is responsible for the rate of calcium reuptake into the lumen of the sarcoplasmic reticulum during diastole, which is in turn a major determinant of the velocity of myocardial relaxation after contraction.13,14
Finally, TH upregulate expression of the sarcoplasmic reticulum calcium-activated ATPase and downregulates phospholamban expression, thereby enhancing myocardial relaxation. Moreover, the improved calcium reuptake during diastole may have a favorable effect on myocardial contractility. Actually, the greater end-diastolic reduction in cytoplasmic concentration of calcium increases the magnitude of the systolic transient of calcium that, in turn, augments its availability for activation of tropo-myosin units. TH lower systemic vascular resistance, increase blood volume, and have inotropic and chronotropic effects on cardiac function; these changes on both the circulation and the heart itself result in increased cardiac output.
A high output circulation state has been described in hyperthyroidism, whereas hypothyroid patients have low cardiac output, decreased stroke volume, decreased vascular volume, and increased systemic vascular resistance. These alterations in cardiac function mediated by TH depend ultimately on the regulation of target genes within the heart and on indirect effects resulting from hemodynamic changes. Moreover, TH play an important role in blood pressure (BP) control but may exert other effects on the cardiovascular system; both hypo and hyperthyroidism may cause cardiovascular dysfunction, increase the response of tissues to the action of the sympathetic system, and this may be a mechanism by which they regulate BP. However, TH can also activate the RAAS without involving the sympathetic nervous system.
A number of cardiovascular changes have been described in thyroid dysfunction, and the right treatment for the condition has shown to be of great benefit. The heart is a major target for TH action, and thyroid dysfunction has profound effects on the heart and cardiovascular system, i.e., changes in cardiac gene expression in the contractile apparatus, the sarcoplasmic reticulum, and the outer myocytic cell membrane. The positive inotropic, dromotropic, and chronotropic heart rates are also increased by TH, and these effects are associated with greater sensitivity of adrenergic and cardiac receptors, as well as increased myosin synthesis.15,16
- A.
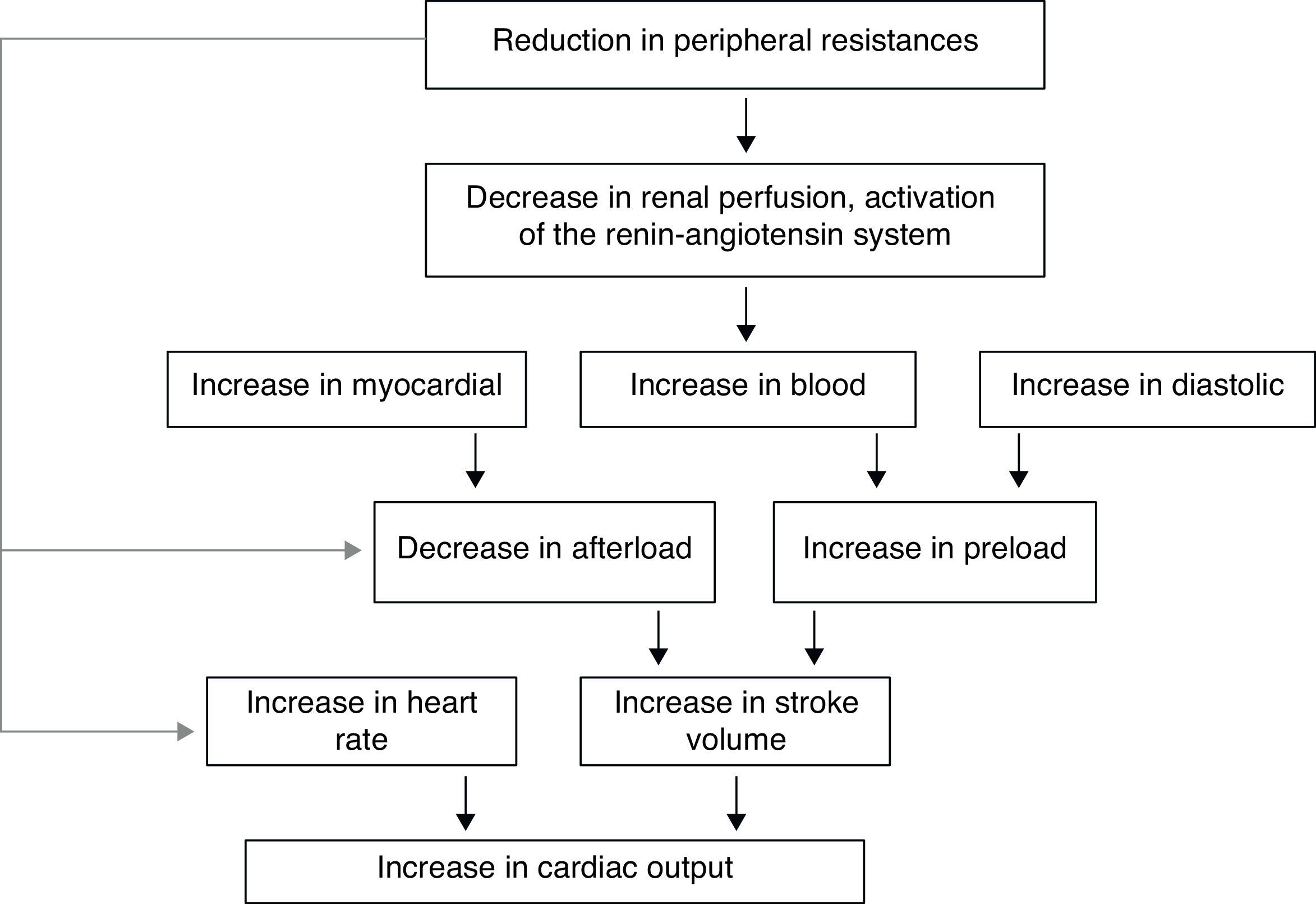
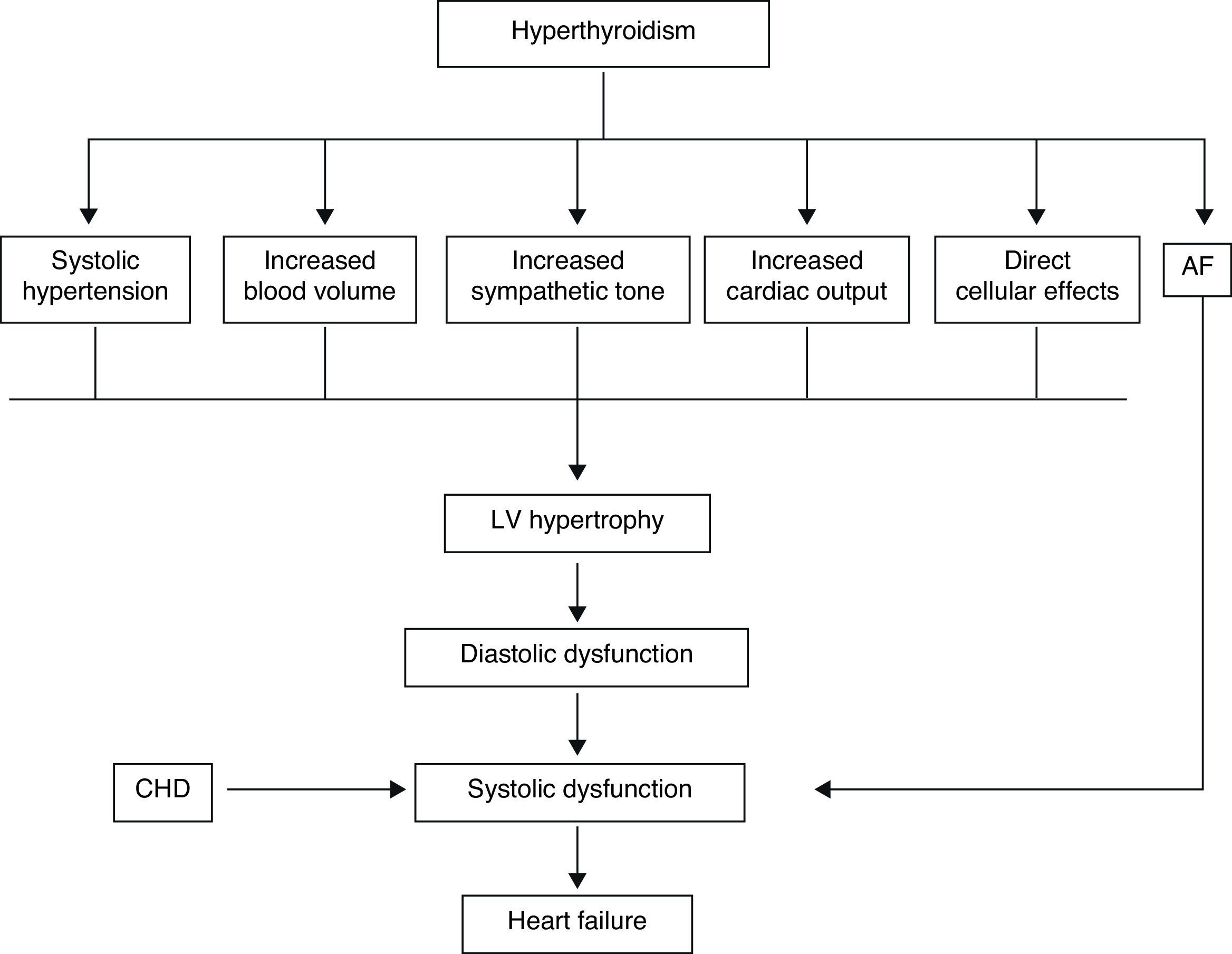
In the hyperthyroid state, TH modulate every component of the cardiovascular system necessary for normal cardiovascular development and function. Excess TH have pronounced cardiovascular manifestations (Table 1). Overall, hyperthyroidism is characterized by an increase in resting heart rate (at least half the patients with hyperthyroidism have sinus tachycardia exceeding 100beats/min) blood volume, stroke volume, myocardial contractility and ejection fraction, and an improvement in diastolic relaxation. An increase in TH level induces resting tachycardia, palpitations are one of the most-common symptoms associated with overt hyperthyroidism, and about 20% of hyperthyroid patients overall have AF. Since symptoms of hyperthyroidism are often non-specific and develop slowly, the latter may be the first clinical manifestation of thyroid dysfunction; and this arrhythmia increases the risk of blood clot formation inside the heart, with embolism and stroke if there is clot dislodgment.17,18 The AF is usually persistent rather than paroxysmal, and is more probable in older patients – perhaps reflecting a reduction in the threshold for this arrhythmia with age. Pulse pressure is widened, cardiac output and sympathetic tone are increased, and a hyperkinetic apex beat and a loud first heart sound are described; in addition, an accentuated pulmonic component of the second sound can be noticed frequently. The increase in chronotropism and batmotropism is probably caused by imbalanced sympathovagal tone due to a relative rather than an absolute adrenergic overdrive. Alterations in the pulse and heart sounds are common, as is also the case with the Means–Lerman “scratch” (mid-systolic and end-tidal murmur heard at the left upper sternal border thought to occur from rubbing of the pericardium against the pleura, which may sound like a pericardial friction rub as seen in pericarditis). Left ventricular (LV) systolic function is consistently increased at rest and the rate of LV chamber relaxation and LV filling is increased.19,20 Additionally, systolic arterial pressure is increased and diastolic arterial pressure is decreased, so that pulse pressure is particularly wider and mean arterial pressure is usually decreased, with a remarkable increase in cardiac output and a notable reduction in peripheral vascular resistance. However, hyperthyroidism has only minor effects on mean arterial blood pressure, because of increases in systolic pressure – caused by increased stroke volume – and decreases in diastolic pressure due to peripheral vasodilatation. The peripheral vascular effects result from a TH-mediated decrease in systemic peripheral resistance, induced by dilating arterioles and by increased metabolic rate in peripheral tissues. As a rule, the total peripheral vascular resistance decreases in thyrotoxicosis, and these alterations may be mediated by changes in non-thyroid hormones which affect the vasculature.21,22 Even though in thyrotoxicosis plasma catecholamines are unchanged or low, the β-adrenergic receptor density is altered in a time- and tissue-dependent manner, raising tissue sensitivity to catecholamines. The rapid use of oxygen, increased production of metabolic end-products, and relaxation of arterial smooth muscle fibers by TH cause peripheral vasodilatation, leading to a reduction in peripheral vascular resistance, and contributing to a further increase in heart rate; concomitantly there is a selective blood flow increase in some sites such as the skin, skeletal muscles and heart, and a fall in diastolic pressure with a simultaneous widening of pulse pressure. The vasodilatation present and the lack of an increase in renal blood flow generate a reduction in renal perfusion pressure, with activation of the RAAS, which increases sodium retention and blood volume. These changes result in preload increase and afterload reduction, leading to a significant increase in stroke volume.23,24 A higher cardiac preload may trigger secretion of atrial natriuretic peptide (ANP); however, it is suggested that TH-induced myocardial ANP secretion in healthy subjects is not the result of a direct action on the myocardium, but rather the result of an indirect modification in cardiovascular hemodynamic leading to increased atrial stretch. Therefore, hyperthyroidism is characterized by a high cardiac output state with a remarkable increase in heart rate and cardiac preload and a reduction in peripheral vascular resistance, resulting in hyperdynamic circulation (Fig. 2). Moreover, increased pressure in the left atrium increases pressure in the pulmonary veins, and this in turn causes reflex contraction of the arterioles in the lesser circulation (Kitaev's reflex) due to stimulation of baroreceptors. Spasm in the arterioles produces a significant increase in pulmonary artery pressure to intensify the load on the right ventricle, which needs contact with a greater force in order to eject blood into the pulmonary trunk, leading to the increase of pulmonary resistance and pulmonary hypertension. Several mechanisms have been suggested in the pathogenesis of pulmonary artery hypertension in patients with hyperthyroidism, including an autoimmune process associated with endothelial damage or dysfunction, increased cardiac output and increased metabolism of intrinsic pulmonary vasodilating substances, all these with normal pulmonary artery resistance. Although the mechanism is uncertain, the reversal of pulmonary artery hypertension following restoration to a euthyroidism state supports a causal relationship. A possible explanation includes an influence of TH, which affects growth and maturation of vascular cells, and enhanced catecholamine sensitivity, causing pulmonary vasoconstriction. Therefore, pulmonary artery hypertension should be considered in hyperthyroid patients with dyspnea.
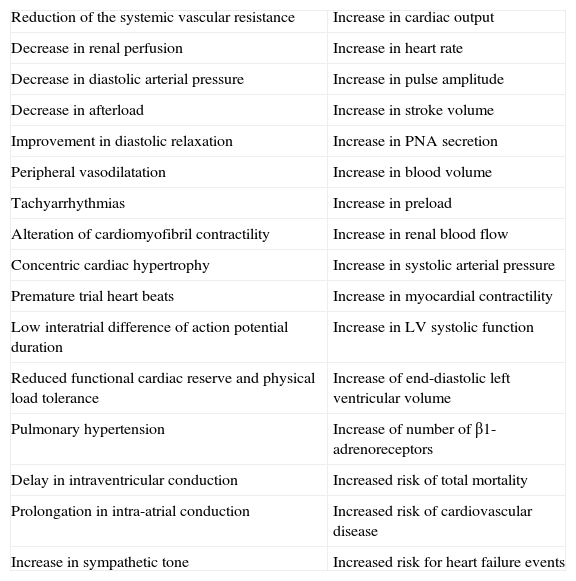
Table 1.Cardiac and hemodynamic consequences of hyperthyroidism.
Reduction of the systemic vascular resistance Increase in cardiac output Decrease in renal perfusion Increase in heart rate Decrease in diastolic arterial pressure Increase in pulse amplitude Decrease in afterload Increase in stroke volume Improvement in diastolic relaxation Increase in PNA secretion Peripheral vasodilatation Increase in blood volume Tachyarrhythmias Increase in preload Alteration of cardiomyofibril contractility Increase in renal blood flow Concentric cardiac hypertrophy Increase in systolic arterial pressure Premature trial heart beats Increase in myocardial contractility Low interatrial difference of action potential duration Increase in LV systolic function Reduced functional cardiac reserve and physical load tolerance Increase of end-diastolic left ventricular volume Pulmonary hypertension Increase of number of β1-adrenoreceptors Delay in intraventricular conduction Increased risk of total mortality Prolongation in intra-atrial conduction Increased risk of cardiovascular disease Increase in sympathetic tone Increased risk for heart failure events - B.
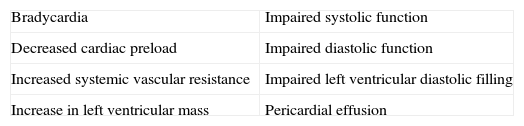
In the hypothyroid state, a deficiency in TH compromises the function of the cardiac muscle by decreasing the activity of enzymes involved in the regulation of calcium uptake and the expression of several contractile proteins in cardiomyocytes, resulting in lower heart rate and weakening of myocardial contraction and relaxation. The most obvious effect of TH deficiency on the heart is a prolongation of both systolic and early diastolic time characteristics.25,26 In the hypothyroid heart, in contrast to congestive heart failure, pulmonary pressure is not increased; hypothyroid patients have reduced cardiac output, stroke volume and plasma volume. Even though hypothyroidism causes fewer cardiovascular symptoms and signs, it is associated with bradycardia, increased vascular resistance, narrow pulse pressure and mild hypertension. Circulation time is prolonged, but right and left heart filling pressures are usually within normal limits, unless they are elevated by pericardial effusion. Venous pressure is normal, but peripheral resistance is increased; there is a redistribution of blood flow with marked reduction in cerebral, renal and cutaneous flow. Cardiac oxygen consumption is reduced even further than what is anticipated from the decreased work load, making for an energy-efficient state of cardiac contraction. However, congestive heart failure has been described in severely hypothyroid patients without underlying heart disease. Measurements of isovolumetric relaxation time reveal a prolongation of this interval.27,28 In addition, there is prolongation of the pre-ejection period and an increased pre-ejection period to LV ejection time ratio (Table 2). Myocardial work efficiency is lower in normal subjects than other sick persons. Angina pectoris, diastolic hypertension, atrioventricular blocks, and pericarditis are major cardiovascular complications in a hypothyroid state. Diastolic dysfunction both at rest and on exertion is the most uniformly found cardiac abnormality in patients with hypothyroidism; LV diastolic function is altered, with a slowed myocardial relaxation and impaired early ventricular filling. This is frequently associated with a fluctuating impairment in LV systolic function even at a very early stage. LV asynchrony is defined as deterioration of the simultaneous contraction of corresponding cardiac segments; as a result, delayed activation of some ventricular segments leads to uncoordinated contraction. LV asynchrony may affect diastolic and systolic functions, exercise capacity, prognosis, quality of life, and symptoms of heart failure, worsening the heart failure. LV systolic function is marginally subnormal, with slightly lower ejection fraction and stroke volume values.29,30 Preload is reduced, with a subnormal cardiac output. High cholesterol levels are an additional risk for the development of atherosclerosis. Alterations in the pulse and peripheral vasoconstriction may be observed, such as prolongation of the QRS complex and the QT interval (the QT interval reflects traditional electrocardiographic parameter of the duration of ventricular repolarization) with an increased risk of developing ventricular tachyarrhythmias. QT dispersion is the inter-lead variability of the QT interval on surface electrocardiogram, reflecting regional variations in myocardial repolarization. Increased QT dispersion has been linked to the occurrence of malignant ventricular arrhythmias and sudden cardiac death; clinical observations show that ventricular arrhythmias and sudden death are uncommon in hypothyroidism, despite the marked prolongation of the QT interval. However, increased QT dispersion in hypothyroidism may facilitate ventricular arrhythmias with hypokalemia, hypomagnesemia, long QT syndrome, and sudden cardiac death.
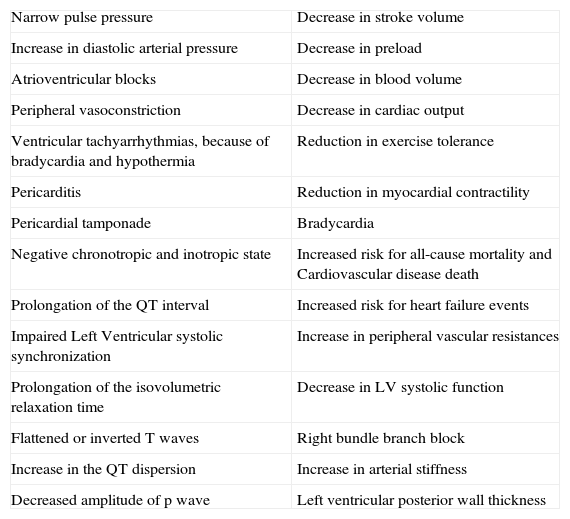
Table 2.Cardiovascular and hemodynamic changes in a hypothyroid state.
Narrow pulse pressure Decrease in stroke volume Increase in diastolic arterial pressure Decrease in preload Atrioventricular blocks Decrease in blood volume Peripheral vasoconstriction Decrease in cardiac output Ventricular tachyarrhythmias, because of bradycardia and hypothermia Reduction in exercise tolerance Pericarditis Reduction in myocardial contractility Pericardial tamponade Bradycardia Negative chronotropic and inotropic state Increased risk for all-cause mortality and Cardiovascular disease death Prolongation of the QT interval Increased risk for heart failure events Impaired Left Ventricular systolic synchronization Increase in peripheral vascular resistances Prolongation of the isovolumetric relaxation time Decrease in LV systolic function Flattened or inverted T waves Right bundle branch block Increase in the QT dispersion Increase in arterial stiffness Decreased amplitude of p wave Left ventricular posterior wall thickness Other findings are: incomplete or complete right bundle branch block, decreased p wave amplitude, diffuse flattening or inversion of T waves together with a generalized low voltage of all the complexes. The T wave is dome-shaped and partially obliterates the ST segment (“the mosque sign”). Isolated myxedema may cause heart failure, pericardial effusion (PE) and pericardial tamponade (PT), especially in subjects with profound T4 deprivation; PE in hypothyroidism is common and the mechanisms of myxedematous PE are increased permeability of capillaries with subsequent leakage of protein rich fluid into the interstitial space, impaired lymphatic drainage, and salt and water retention; nevertheless, an effusion which causes cardiac tamponade is rarely seen. PT in hypothyroid patients with PE is attributed to the slow accumulation of fluid and the remarkable compliance of the pericardium.
The heart in overt myxedema is often flabby, and grossly dilated. Classic findings of overt myxedema are: cardiac enlargement, dilatation, significant bradycardia, weak arterial pulses, hypotension, distant heart sounds, low EKG voltage, non-pitting edema and evidence of congestive heart failure.
There is a relationship between hypothyroidism and coronary artery disease, either because of the presence of a negative chronotropic and inotropic state or the presence of hypercholesterolemia and hypertension, with an increased risk of atherogenesis; but otherwise, thyroid hormones are powerful regulators of vasculature in the adult myocardium; therefore, a low free T3 state would inhibit neovascularization in cardiac tissue after acute myocardial infarction, which would accelerate cardiac pathologic remodeling and heart failure, leading to short-term and long-term adverse cardiac events.31,32

Besides its metabolic and thermoregulatory tissue effects, TH regulate cardiac performance by acting on the heart and vascular system. Hyperthyroid patients can manifest findings of congestive heart failure in the absence of prior cardiac injury. Diastolic and systolic functions are clearly modified by TH; ventricular contractile function is also altered by changes in the hemodynamic pattern, secondary to TH effects on peripheral vascular tone. TH equilibrium preserves positive ventricular–arterial coupling, leading to an adequate balance for cardiac work. Hemodynamic alterations due to hyperthyroidism decrease myocardial contractile reserve and do not allow further increases in ejection fraction and cardiac output on exertion, probably because of the inability to reduce the already low peripheral vascular resistance, while constriction of venous vessels increases. The resulting decrease in peripheral vascular resistance activates the RAAS, leading to retention of sodium and fluid.33,34
The forced increase in preload and total blood volume increases cardiac work and stimulates the development of myocardial hypertrophy. However, the increase in preload and blood volume leads to a rise in ventricular filling pressure, and to a moderate degree of pulmonary and peripheral congestion. Cardiac output is augmented by a higher heart rate and increased stroke volume, facilitating the return of blood to the heart. These changes result in increased mean circulatory filling pressure, which promotes retrograde blood flow to the right atrium. However, TH also increase erythropoiesis, and the net effect is an increase in total blood volume and stroke volume.
The transcriptional effects lead to increased contractility through effects on the release and uptake of sarcoplasmic reticular calcium and phosphorylation of phospholamban. The non-transcriptional effects are induced by the effect of TH on various ion channels.35,36 All these cardiac effects, along with low peripheral vascular resistance and increase in total blood volume, lead to a high cardiac output state (often called “high-output heart failure”). However, “heart failure” is not really the appropriate term because cardiac output is increased, although congestive circulation is present (Table 3). The tachycardia observed in hyperthyroidism appears to be due to a combination of increased rate of diastolic depolarization and decreased duration of the action potential in the sinoatrial nodal cells. On the other hand, the development of “high-output heart failure” in hyperthyroidism may be due to “tachycardia-mediated cardiomyopathy” (TMC). A high cardiac output has been described as being 8L/min or a cardiac index >3.9L/min/m2. TMC is defined as secondary ventricular dysfunction due to chronic tachycardia, which is fully or partially recoverable after heart rate normalization. The diagnosis should be suspected in patients with compromised ventricular function in the course of a ventricular or supraventricular tachycardia. The diagnosis can only be established with the recovery of ventricular function once the tachycardia and the thyrotoxic state are under control.37,38 While TMC usually presents with significant cardiac enlargement, reduced ventricular wall thickness, and impaired ventricular contraction similar to dilated cardiomyopathy, the cardiac abnormalities normalize with control of the tachyarrhythmia and heart failure.
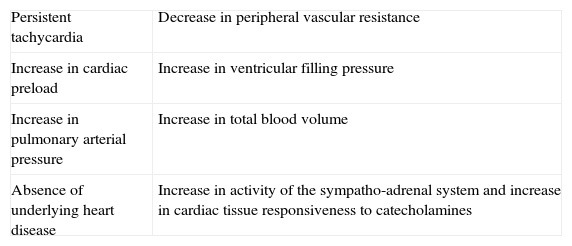
Characteristics that define “high-output heart failure” in hyperthyroidism.
| Persistent tachycardia | Decrease in peripheral vascular resistance |
| Increase in cardiac preload | Increase in ventricular filling pressure |
| Increase in pulmonary arterial pressure | Increase in total blood volume |
| Absence of underlying heart disease | Increase in activity of the sympatho-adrenal system and increase in cardiac tissue responsiveness to catecholamines |
Actually, it has been proposed that cardiovascular effects of hyperthyroidism, i.e., tachycardia, increased cardiac output, systolic hypertension, and myocardial contractility are the result, not only of increased activity of the sympatho-adrenal system, but also of increased cardiac tissue responsiveness to catecholamines, with upregulation of beta adrenergic receptors (Figs. 3 and 5).



The term “thyrotoxic cardiomyopathy” defines myocardial damage caused by the toxic effects of abundant TH, resulting in altered energy production by myocytes (oxidative phosphorylation, glycolysis), intracellular metabolism (protein synthesis) and myofibril contractile function. The main manifestations are left ventricular hypertrophy, heart rhythm disturbances – usually, atrial fibrillation – dilation of the heart chambers and heart failure, pulmonary hypertension, and diastolic dysfunction. It is not known whether cardiomyopathy in hyperthyroidism is secondary to direct toxic effects of excess thyroid hormone, whether it results from the hyperdynamic or high-output stress caused by the thyroid hormone, or whether it is caused by a combination of both. However, cardiomyopathy caused by hyperthyroidism has been shown to be reversible in adults with anti-thyroid therapy. Factors which may play a role in recovery are β-blocker administration and high T3 serum levels.
Three stages of thyrotoxic cardiomyopathy are defined:
- 1.
Hyperkinetic: In which left ventricular function is preserved, but left ventricular ejection fraction does not increase with exertion.
- 2.
Normokinetic: It is a compensatory stage, where there is a reversible myocardial hypertrophy with preserved cardiac output.
- 3.
Hypokinetic: It is a decompensation stage, where there is low cardiac output and stroke volume, reversible or irreversible heart chamber hypertrophy and dilation.
Moreover, “right ventricular heart failure” may result from right ventricular volume overload, due to the increased blood volume and venous return. It is characterized by right ventricular dilation, enlargement of the tricuspid valve annulus and tricuspid insufficiency, and is frequently associated with pulmonary hypertension.39,40
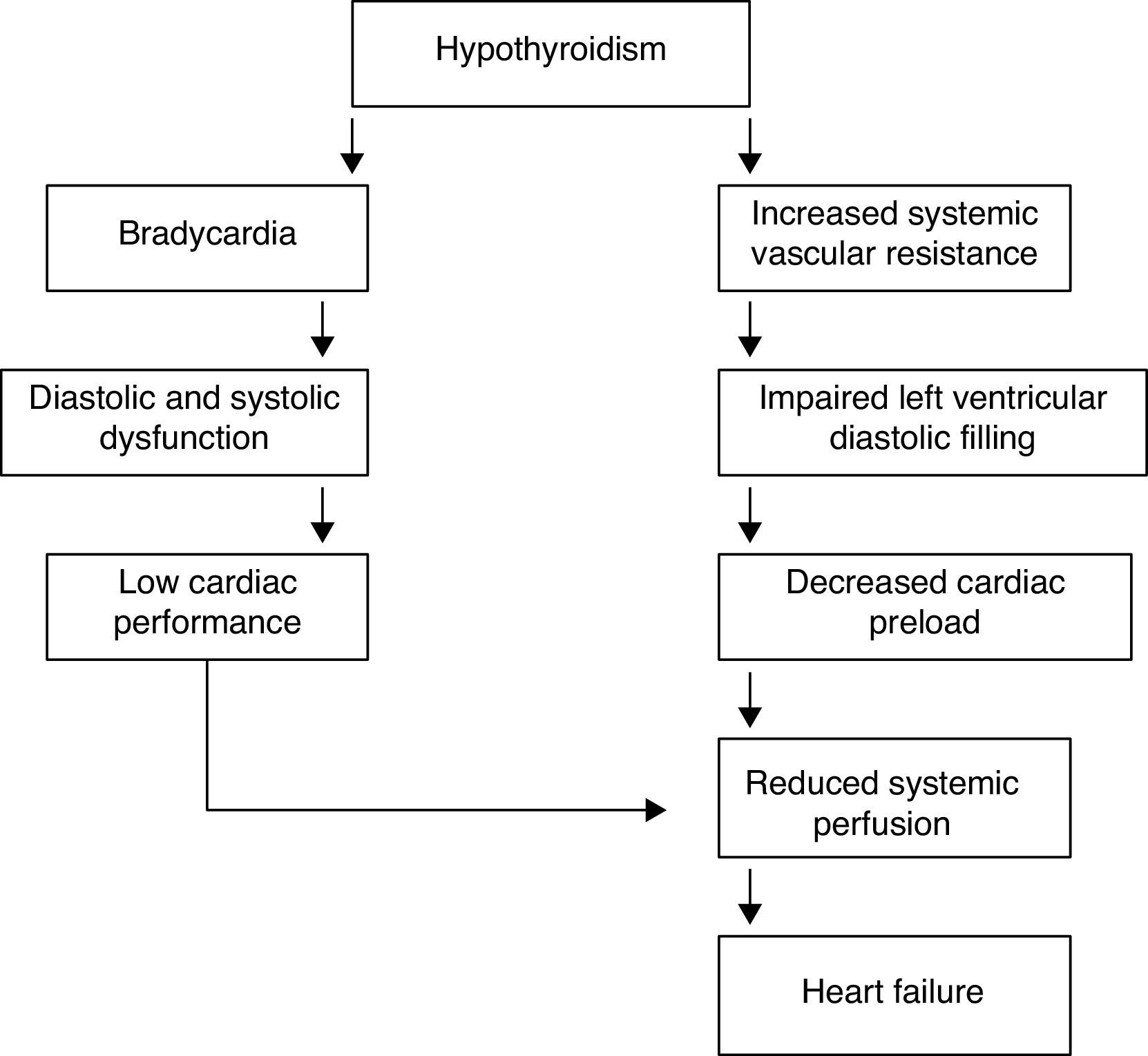
Heart failure in hypothyroidismCardiac changes in hypothyroidism are the complete opposite of those occurring in thyrotoxicosis. The most prominent findings are the decrease in cardiac output and cardiac contractility, diastolic hypertension, increased systemic vascular resistance, and rhythm disturbances; systolic and diastolic functions are reduced at rest and during exercise. TH deficit decreases tissue thermogenesis and increases resistance in peripheral arterioles through the direct effect of T3 on vascular smooth muscle cells. Cardiac preload is decreased due to the impaired diastolic function and to the decreased blood volume (Table 4). Cardiac afterload is increased and chronotropic and inotropic functions are reduced, resulting in a decrease in cardiac output.41,42 The physiological chronotropic response and normal tension of the heart muscle in diastolic phase depend on the proper expression of T3 in the heart cells and its stimulating influence on Na+–K+-ATPase and Ca2+-ATPase in the endoplasmic reticulum. The isovolumetric relaxation phase of diastolic function slows down, just like the contraction velocity during systole, and there is chamber dilatation and impaired myocardial blood flow (Fig. 4).
Dilated cardiomyopathy (DCM) is a heart muscle disorder defined by the presence of a dilated and poorly functioning left ventricle in the absence of abnormal loading conditions (hypertension, valve disease) or ischemic heart disease sufficient to cause global systolic impairment; in hypothyroidism, although cardiac output is reduced, heart failure is relatively rare because there is a lower oxygen demand in the periphery. The improvement of the cardiac function after hormonal treatment is an important argument in favor of the implication of hypothyroidism in the genesis of DCM.43,44
Atrial fibrillation and thyroidAF is the most common cardiac complication of hyperthyroidism, occurring in an estimated 10% to 25% of overtly hyperthyroid patients; in comparison 0.4% of the general population has AF, representing an independent risk factor for cardiovascular events.
Prevalence increases with age; so much so that 25% of hyperthyroid patients older than 60 years had AF compared to 5% in patients less than 60 years of age, indicating that age is a major factor in the onset of AF.
The propensity to develop AF may be due to the shortened refractory period of atrial cells and a greater delay in the rectifier potassium current increases between the right atrium and the left atrium, creating a substrate for AF.45,46
Furthermore, TH potentiate the effect of the adrenergic system on the heart, and while catecholamine levels are either normal or decreased in hyperthyroidism, catecholamine action occurs through increased tissue sensitivity due to upregulated transcription of beta-adrenergic receptors and differences in autonomic innervations between atria and ventricles. It is also possible that the sensitivity of atrial or ventricular myocardial cells to TH is different.
Generally, the onset of AF occurs with premature complexes originating from the pulmonary veins, and the persistence of AF requires re-entry; premature complexes occur secondary to automaticity or triggered activity. Hyperthyroidism is associated with reduced vagal activity and reduced heart rate variability; the rapid and irregular heartbeat produced by AF increases the risk of blood clot formation inside the heart.47,48
These clots may eventually become dislodged, causing embolism. Moreover, TH have various effects on coagulation, TH excess is associated with coagulation abnormalities, such as shortened activated partial thromboplastin time, increased fibrinogen levels, and increased factor VIII and factor X activity in patients in sinus rhythm with thyrotoxicosis; even so, the best evidence-based study did not find a trend toward increased embolic risk.
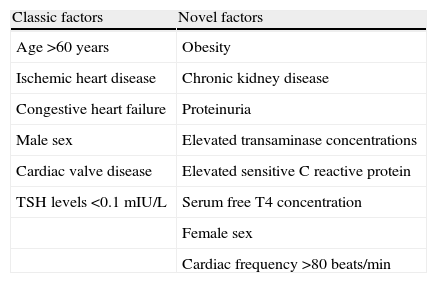
AF alters atrial electrical and structural properties in a way that promotes its own maintenance; this increases the risk of recurrence and may alter the response to antiarrhythmic drugs. The risk factors for AF in patients with hyperthyroidism are similar to those in the general population (age, ischemic heart disease, congestive heart failure, male sex and valvular heart disease). However, other factors have been associated with the presence of AF in hyperthyroidism (Table 5) including obesity, chronic kidney disease (which is a powerful predictor of new-onset AF in hypertensive patients, independently of LV hypertrophy and left atrial dilatation), proteinuria, female sex, serum free T4 concentration, and elevated transaminase concentrations.49,50
Risk factors for AF in patients with hyperthyroidism.
| Classic factors | Novel factors |
| Age >60 years | Obesity |
| Ischemic heart disease | Chronic kidney disease |
| Congestive heart failure | Proteinuria |
| Male sex | Elevated transaminase concentrations |
| Cardiac valve disease | Elevated sensitive C reactive protein |
| TSH levels <0.1mIU/L | Serum free T4 concentration |
| Female sex | |
| Cardiac frequency >80beats/min |
Moreover, it has been suggested that high sensitive C reactive protein, an indicator of inflammation, free T4, and left atrial diameter are associated with the development of AF in patients with hyperthyroidism.
Moreover, hypothyroidism is associated with bradycardia, decreased variability in heart rate, and has been associated with a lower risk of AF compared with euthyroid patients.51,52
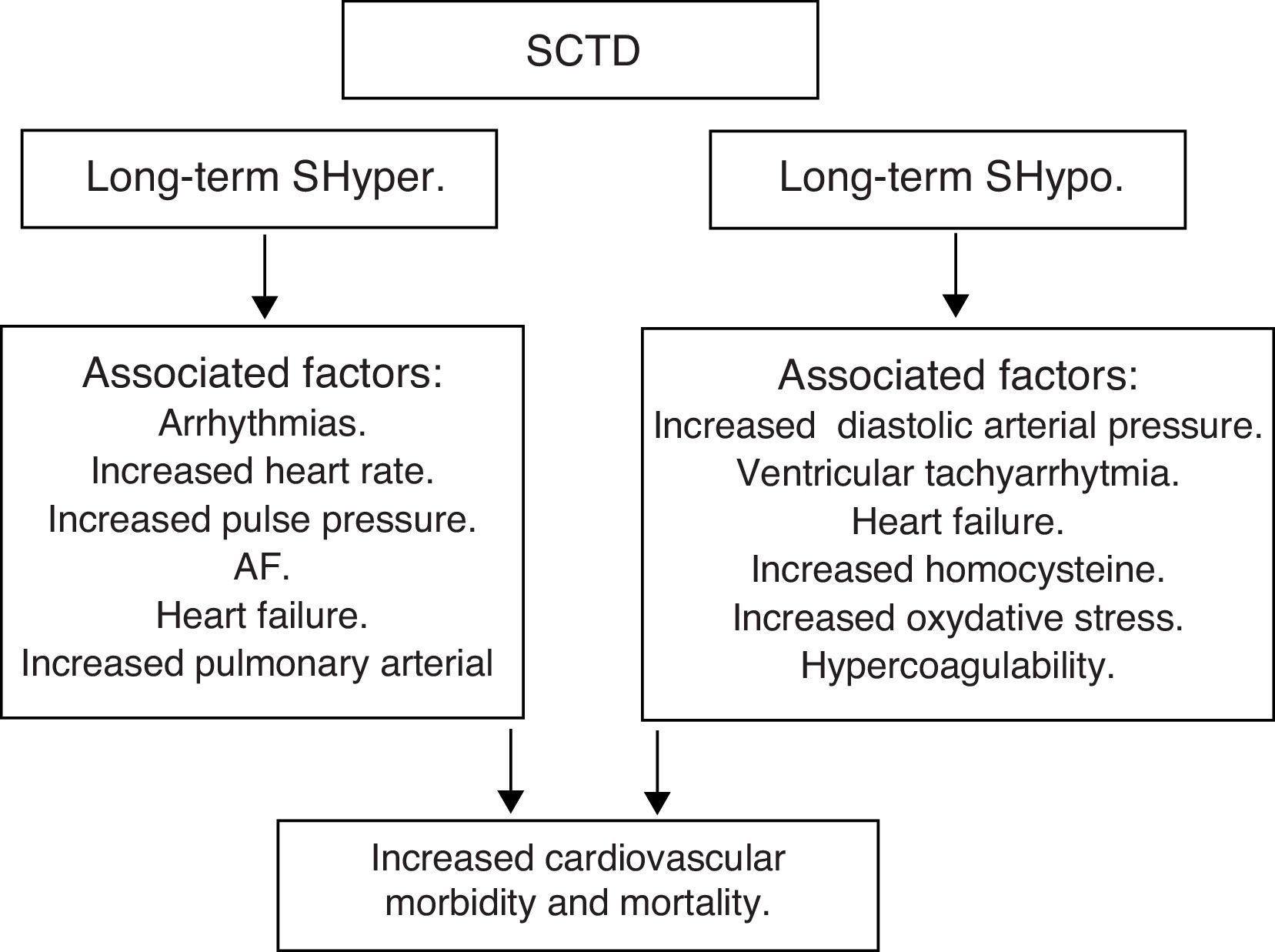
Subclinical thyroid dysfunction (SCTD) and cardiovascular disease and mortalityAlthough it is recognized that patients with SCTD may have subtle symptoms of thyroid dysfunction, the definition is purely a biochemical one: SCTD is defined as serum free T4 and total or free T3 levels within their respective reference ranges in the presence of abnormal serum TSH levels. Serum TSH is undetectable or low in subclinical hyperthyroidism (SHyper), and it is increased in subclinical hypothyroidism (SHypo). It is a common finding in the growing population of elderly patients, occurring in 10–15% among those aged 65 and older.53,54
Controversy persists about whether screening and treating subclinical thyroid dysfunction is warranted.
SHypo has been associated with elevated cholesterol levels and increased risk for atherosclerosis. SHyper has been associated with cardiovascular and total mortality. The relationship between SHypo and coronary heart disease (CHD) seemed to differ among studies that involved middle-aged versus elderly participants, with studies whose samples had a mean age younger than 65 years showing increased risk for CHD.
A meta-analysis showed that SCTD might represent a potentially modifiable – albeit modest – risk factor for CHD and mortality. Potential mechanisms for the associations with cardiovascular diseases among adults with SHypo include elevated cholesterol levels, increased homocysteine and oxidative stress, insulin resistance, increased systemic vascular resistance, arterial stiffness, endothelial dysfunction, and activation of thrombosis and hypercoagulability. The common denominator in the various studies is the heterogeneity among individual studies that used different TSH cutoffs, different confounding factors for adjustment, and varying CHD definitions.
The risk of CHD events tends to be higher when TSH is ≥7.0mUI/L, being more evident when the value is ≥10mUI/L. In summary, for SHypo, combined available data from large prospective cohorts suggest that subclinical hypothyroidism is associated with an increased risk of CHD in those with higher TSH levels. The risk of both CHD mortality and CHD events, but not of total mortality, increases with higher concentrations of TSH, and is significantly elevated in adults with TSH levels of 10mUI/L or greater. Conversely, minimal TSH elevations are not associated with an increased risk of CHD events and CHD mortality.55,56
Moreover, SHyper in age and sex-adjusted analyses was associated with increased total mortality, CHD mortality, CHD events and AF. Risks did not differ significantly by age, sex, or preexisting cardiovascular disease, and were similar after further adjustment for cardiovascular risk factors, with an attributable risk of 14.5% for total mortality and 41.5% for AF. However, heart failure is the leading cause of an increased cardiovascular mortality in both overt hyperthyroidism and subclinical hyperthyroidism.
Risks for CHD mortality and AF (but not other outcomes) were higher for thyrotropin levels under 0.10mUI/L compared with thyrotropin levels between 0.10 and 0.44mUI/L.57,58
In summary, SCTD represents a potentially modifiable risk factor for CHD and mortality, especially when the values of TSH are <0.10mIU/L – Shyper and ≥10.0mIU/L – Shypo (Fig. 6).
Amiodarone and thyroid
Amiodarone is a potent class III anti-arrhythmic drug used in clinical practice for the prophylaxis and treatment of many cardiac rhythm disturbances, ranging from paroxysmal AF to life-threatening ventricular tachyarrhythmias. Amiodarone often causes changes in thyroid function tests mainly related to the inhibition of 5′-deiodinase activity resulting in a decrease in the generation of T3 from T4, with a resulting increase in rT3 production and a decrease in its clearance. However, the use of amiodarone is associated with several side effects owing to its marked lipid affinity. It is highly concentrated in tissues and is linked to a number of adverse effects including photosensitivity, corneal microdeposits, pulmonary toxicity, hepatotoxicity, peripheral neuropathy, lung dysfunction, gynecomastia, ataxia, tremors, peripheral neuropathy, hyperthyroidism and hypothyroidism.59,60
Amiodarone is a benzofuran derivative containing two atoms of iodine per molecule. This amounts to 37.5% of organic iodine by molecular weight, and 10% of the drug's iodine content is released daily as free iodide. Drug doses range from 200 to 600mg daily and treatment releases about 7–20mg of iodide daily, which is about 50–100 fold the optimal daily iodine intake.
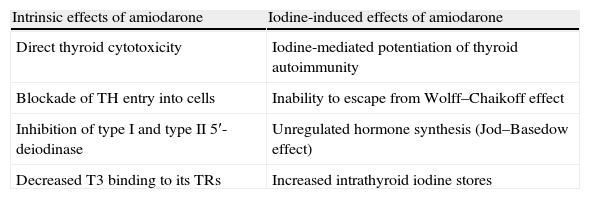
Although the majority of the adverse effects of amiodarone on several organs are due to deposition of the drug in the parenchyma, its effects on the thyroid gland can be divided into two groups: intrinsic effects resulting from the inherent properties of the compound, and iodine-induced effects due solely to the pharmacologic effects of a large iodine load (it has the potential to cause thyroid dysfunction because of its iodine-rich chemical structure) (Table 6).
Amiodarone and its effects on the thyroid gland.
| Intrinsic effects of amiodarone | Iodine-induced effects of amiodarone |
| Direct thyroid cytotoxicity | Iodine-mediated potentiation of thyroid autoimmunity |
| Blockade of TH entry into cells | Inability to escape from Wolff–Chaikoff effect |
| Inhibition of type I and type II 5′-deiodinase | Unregulated hormone synthesis (Jod–Basedow effect) |
| Decreased T3 binding to its TRs | Increased intrathyroid iodine stores |
Amiodarone can lead to both hypothyroidism (amiodarone-induced hypothyroidism – AIH – with a prevalence ranging from 5 to 22%) and, less commonly to hyperthyroidism (amiodarone-induced thyrotoxicosis – AIT – with a prevalence ranging from 2 to 9.6%).
AIT appears to occur more frequently in geographical areas with low iodine intake, whereas AIH is more frequent in iodine-sufficient areas. In contrast to AIT, AIH is slightly more frequent in females (probably due to underlying Hashimoto's thyroiditis).
The most likely pathogenic mechanism is that the thyroid gland is unable to escape from the acute Wolff–Chaikoff effect after an iodine load and to resume normal thyroid hormone synthesis. The large amount of iodide released during the metabolism of amiodarone leads to an adaptive blockage of further thyroidal iodide uptake and TH biosynthesis (Wolff–Chaikoff effect). Although it can be apparent within the first two weeks of treatment, further exposure to iodine leads to normal resumption of TH synthesis. This escape phenomenon from the Wolff–Chaikoff effect helps protect the individual from developing hypothyroidism.61,62
The pharmacological concentrations of iodide associated with amiodarone treatment lead to a protective inhibition of thyroidal T4 and T3 synthesis and release by thyroid within the first two weeks of treatment. After 3 months of amiodarone administration, a steady state is reached, with some hormonal changes persisting indefinitely. Total and free T4 and rT3 remain at the upper end of normal or slightly elevated, and serum T3 levels remain in the low normal range. In contrast, serum TSH levels return to normal after 12 weeks of therapy.
The cause for TSH normalization is presumed to be an increase in the T4 production rate, possibly as a result of increased intrathyroidal iodine stores and escape from the Wolff–Chaikoff effect.63
Two main mechanisms can lead to AIT: iodine-induced hyperthyroidism (type 1 AIT, a form of Jod–Basedow effect, which is identical to that seen in patients with endemic iodine deficient goiter who are given iodide replacement); or destructive thyroiditis with destruction of thyroid follicles resulting in a thyroiditis with excess release of T3 and T4 (type 2 AIT), caused by amiodarone itself and its high iodine content. Type 1 AIT occurs in subjects with an abnormal thyroid (goiter or latent autoimmune disease), with the iodine load triggering autonomous thyroid hormone production.
Type 2 develops in subjects who have an apparently normal gland and may reflect TH release due to direct cytotoxic effects of the drug on thyrocytes. The nature of destructive thyroiditis is that of a self-limiting disease. AIT may develop early during amiodarone treatment or even several months after drug withdrawal. This is because of amiodarone and its metabolites – mainly desethylamiodarone.64,65
Dronedarone is a new anti-arrhythmic drug, which was designed to maintain the potent antiarrhythmic and rate-controlling effects of amiodarone while reducing its toxic effects. Dronedarone is a non-iodinated benzofuran derivative of amiodarone in which the iodine moieties, observed with amiodarone, are substituted with a methyosulfonamide group. However, dronedarone is less lipophilic than amiodarone, with a much shorter half-life (24h) than amiodarone (several weeks); it is also extensively metabolized primarily by the cytochrome P450 3A4 system and excreted in the bile with minimal renal excretion. Dronedarone does not appear to cause any of the thyroid, pulmonary and neurological adverse effects observed with amiodarone.66–68
ConclusionsThyroid dysfunction causes remarkable cardiovascular derangements.
TH has numerous effects on the cardiovascular system in physiological conditions which are mediated mainly by intracellular receptors, but also through non-genomic pathways. On the basis of the understanding of the cellular mechanisms of TH action on the cardiovascular system, it is possible to explain the mechanism of cardiac output, cardiac contractility, blood pressure, vascular resistance, and rhythm disturbances that result from thyroid dysfunction.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflict of interestThe authors declare that there is no conflict of interest that could bias the impartiality of this review.