




Los fibratos o derivados del ácido fíbrico se utilizan en la farmacoterapia de diversos tipos de hiperlipemias desde hace más de 3 décadas. El fármaco cabeza de serie del grupo, hoy ya en desuso, es el clofibrato o clorofenoxiisobutirato de etilo. Su hidrólisis en plasma proporciona la forma activa, el ácido clofíbrico, forma halogenada del ácido fenoxiisobutírico o ácido fíbrico, de aquí la denominación de este grupo farmacológico. A partir de la molécula inicial de clofibrato, se diseñaron diversas moléculas que contenían en su estructura el ácido fíbrico, como etofibrato, binifibrato, etc. Hoy día se utilizan fibratos de segunda generación, con estructuras químicas más o menos relacionadas con el ácido fíbrico, como son el bezafibrato, el fenofibrato y el gemfibrozilo.
Los fármacos de este grupo poseen un marcado efecto hipotrigliceridémico (hasta un 50% de reducción en la concentración de triglicéridos plasmáticos), incrementan acusadamente el colesterol unido a lipoproteínas de alta densidad (cHDL) (entre un 5 y un 20%) y manifiestan un efecto variable en las concentraciones de colesterol unido a lipoproteínas de baja densidad (cLDL), aunque, en cualquier caso, siempre modifican el patrón de LDL circulantes hacia formas de baja densidad y gran tamaño, que presentan una menor aterogenicidad1. Por todo ello, se utilizan en el tratamiento de las hipertrigliceridemias, hiperlipemias mixtas y, especialmente, en el control de la dislipemia asociada a la diabetes y el síndrome metabólico.
Mecanismo de acción y efectos farmacológicos
Fue necesario que transcurrieran prácticamente 2 décadas desde el comienzo de la utilización terapéutica de los fibratos para que, al inicio de la década de los años noventa, se identificara su mecanismo de acción. Se conocía que los fibratos administrados a roedores, principalmente rata y ratón, inducían de forma intensa la proliferación de diversos orgánulos celulares en los hepatocitos e incrementaban las actividades enzimáticas relacionadas con ellos. Dado que los peroxisomas son, con diferencia, los orgánulos más afectados por el fenómeno proliferativo, tanto los fibratos como otros compuestos con propiedades similares recibieron el nombre genérico de proliferadores peroxisómicos2. En 1990, Isseman y Green3 identificaron el primer receptor nuclear activado por proliferadores peroxisómicos. Dicho receptor es el que hoy conocemos como receptor activado por proliferadores peroxisómicos alfa o PPAR* (NR1C1) y es causa de la aparición de la mayoría de los efectos farmacológicos atribuibles a los fibratos. En los años siguientes se han identificado otros 2 receptores pertenecientes a la misma subfamilia, los receptores PPARß/* (NR1C2) y PPAR* (NR1C3), de los que este último es el receptor diana de un nuevo grupo terapéutico en el tratamiento de la diabetes mellitus tipo 2, las tiazolidindionas o glitazonas4.
Los receptores PPAR se clasifican dentro de la subfamilia de receptores nucleares de clase II5. En este grupo se encuentran receptores de hormonas clásicas, como las hormonas tiroideas (TR) y la vitamina D (VDR), y receptores considerados huérfanos hasta hace pocos años, como es el caso de los receptores de los derivados de la vitamina A, los ácidos transretinoicos (RAR), receptores activados por oxisteroles (LXR) y receptores activados por ácidos biliares (FRX). Una gran mayoría de estos receptores controlan el metabolismo de los ácidos grasos y el colesterol y se caracterizan por poseer una serie de rasgos comunes6,7:
Siempre actúan en forma de heterodímeros con el receptor del ácido 9-cisretinoico (RXR), uniéndose siempre el RXR en la zona 5' del HRE. Normalmente, cuando el RXR forma parte de un heterodímero, pierde su capacidad de activación de la transcripción, excepto en el caso del heterodímero con PPAR, en el que se produce una potenciación de la transcripción en concomitancia de agonistas para el RXR y PPAR.
La mínima zona de consenso para el HRE es la secuencia 5'-AGGTCA-3', presentada en la denominada "norma 1-5", que implica que esta secuencia se presenta en forma de una repetición directa separada por 1 (PPAR), 2 (RXR), 3 (VDR), 4 (TR, LXR) o 5 (RAR) nucleótidos. Esta situación es la más común, aunque se puede encontrar repeticiones invertidas (FXR) o incluso evertidas.
Los PPAR, además de ser heterodímeros del receptor RXR, interaccionan con otras proteínas denominadas cofactores de expresión, los denominados correpresores y coactivadores de expresión8,9. Entre los factores correpresores, los más comunes son las proteínas SMRT (silent mediator for RARs and TRs) y N-CoR (nuclear receptor corepresor). Estos correpresores interaccionan con el receptor libre inhibiendo la actividad transcripcional basal del receptor. La unión con el ligando específico induce la disociación del correpresor. Por el contrario, los coactivadores facilitan la interacción entre la zona AF-2 del PPAR y la maquinaria de transcripción una vez se ha producido la activación del PPAR por unión a su ligando específico; las proteínas SCR-1 (steroid receptor coactivator-1) y p300 se han identificado como coactivadores de los PPAR. De hecho, hoy día parece evidente que no todos los ligandos de PPAR seleccionan por igual el mismo conjunto de proteínas coactivadoras, de forma que, aunque existe un núcleo de efectos comunes a los ligandos de una determinada isoforma de PPAR, cada ligando individual puede presentar efectos particulares y diferenciados del resto.
Independientemente de la activación típica por ligando, existen evidencias de que los PPAR pueden ser activados por otros mecanismos alternativos. Existen datos sobre la posible modulación de la actividad PPAR mediante procesos de fosforilación/ desfosforilación, aunque son bastante contradictorios. Por ejemplo, la fosforilación de los PPAR por acción de cinasas como la proteincinasa activada de mitógenos (MAPK) puede conducir a un incremento o a una reducción de la actividad transcripcional de los PPAR, dependiendo del tipo de señal fisiológica que origina el estímulo10,11.
El PPAR* se expresa mayoritariamente en el hígado y, en menor medida, en corazón, riñón, músculo esquelético e intestino; en el caso particular de la rata, también se encuentra abundantemente en el tejido adiposo marrón12,13. En todos estos tejidos, el PPAR* se encuentra directamente implicado en el control de la expresión de genes que codifican proteínas y enzimas clave en el metabolismo energético, especialmente en el catabolismo de los ácidos grasos.
Hoy conocemos diversos ligandos endógenos de los PPAR; todos ellos son ácidos grasos o derivados metabólicos de éstos. Todos los ácidos grasos poliinsaturados se comportan como ligandos del PPAR a concentraciones fisiológicas y presentan, en general, una mayor afinidad para la isoforma PPAR*14,15. Entre los productos del metabolismo de los ácidos grasos cabe destacar el leucotrieno B4 y el ácido 8(S)-HETE, que se comportan como ligandos selectivos de la isoforma PPAR*16. Como ligandos sintéticos caben destacar el ácido ETYA y los fibratos hipolipemiantes (clofibrato, bezafibrato, gemfibrozilo, Wy-14.643, etc.), como ligandos selectivos del PPAR*; las tiazolidindionas hipoglucemiantes (troglitazona, rosiglitazona, pioglitazona, etc.) son ligandos selectivos del PPAR*.
La activación transcripcional de diversos genes por PPAR*, especialmente en el tejido hepático, causa la manifestación de los efectos hipolipemiantes clásicos de los fibratos17. Esto se debe, básicamente, a la modificación de la expresión de 3 conjuntos de genes bien diferenciados:
Incremento de la expresión de genes como ACO y L-CPT-I, que codifican enzimas limitantes de los procesos de oxidación de ácidos grasos en peroxisomas (acil-CoA oxidasa) y mitocondrias (L-carnitinapalmitoil-transferasa-I)18,19. El mayor catabolismo de ácidos grasos conduce a una reducción de la síntesis de triglicéridos y a una menor producción de lipoproteínas de muy baja densidad (VLDL) por el tejido hepático.
Incremento en la expresión de la lipoproteinlipasa hepática y reducción en la expresión de la apolipoproteína C; ambos efectos conducen a un aumento de la actividad lipoproteinlipasa y, por consiguiente, a un aumento del catabolismo de las VLDL y una reducción de los triglicéridos hepáticos. Paralelamente, la reducción del tiempo de permanencia de las VLDL en circulación y la aceleración de su transformación en LDL favorece la formación de lipoproteínas de gran tamaño y baja densidad, con un potencial aterogénico reducido20.
Incremento en la expresión de las apolipoproteínas (Apo) AI y Apo-II y transportadores de colesterol, como CLA-1 y ABCA1. En su conjunto, estas modificaciones conducen al incremento de las concentraciones plasmáticas de HDL y al aumento del transporte inverso de colesterol21.
En la actualidad, se considera que la aterosclerosis es un proceso inflamatorio crónico, con aparición de manifestaciones clínicas agudas cuando se produce la rotura de una placa ateromatosa, trombosis sobreañadida y la consiguiente isquemia tisular22-24. PPAR* y PPAR* se expresan en diversos tipos celulares relevantes tanto para el sistema inmunitario como para el desarrollo de la aterosclerosis; entre ellos encontramos linfocitos, monocitos, macrófagos, células endoteliales, células musculares lisas, etc.25. De forma similar a lo ocurrido con las estatinas, el análisis de los resultados del estudio VA-HIT indica que la reducción observada en la morbilidad y la mortalidad de causa cardiovascular en el grupo tratado con gemfibrozilo, un reconocido agonista PPAR*, es superior a la atribuible a los modestos cambios detectados en las concentraciones plasmáticas de triglicéridos y cHDL26. Por tanto, sería previsible esperar la existencia de otros efectos "pleiotrópicos", no directamente relacionados con el clásico efecto hipolipemiante, derivados de la activación de los receptores PPAR. Entre estos posibles efectos pleiotrópicos, la modulación de la actividad inflamatoria/sistema inmunitario mediante la activación de los diversos subtipos de receptores PPAR ha conseguido, en los últimos años, una sólida base experimental en que sustentarse27-29.
La primera evidencia de la implicación de los PPAR en el proceso inflamatorio fue proporcionada por un estudio que demostraba que en ratones con una ausencia del gen PPAR* se prolongaba la duración de la respuesta inflamatoria16. PPAR* controla la degradación del eicosanoide proinflamatorio LTB4 a través de los procesos de hidroxilación omega y betaoxidación peroxisómica30. Devchand et al16 confirmaron que este mediador de la inflamación se unía a PPAR* e inducía la transcripción de genes implicados en la oxidación omega y beta peroxisómica en el hígado, lo que da lugar a un incremento de su propio metabolismo. Puesto que PPAR* controla la degradación de mediadores lipídicos de la inflamación, estos resultados indicaban que este factor de transcripción podía regular la duración del proceso inflamatorio. Utilizando ratones con una ausencia del gen PPAR* se demostró una mayor duración del proceso inflamatorio respecto a los ratones que expresaban este receptor, lo que confirmó la participación de PPAR* en el control de la inflamación. Desde el trabajo pionero de Devchand et al16 en 1996, se han publicado numerosos trabajos que confirman la actividad antiinflamatoria de este receptor. La actividad PPAR* inhibe, en diversos modelos experimentales, la producción de marcadores proinflamatorios en células endoteliales, como endotelina 1, ICAM-1 (intracellular adhesion molecule-1) y VCAM-1 (vascular cell adhesion molecule-1), y macrófagos, como la metaloproteinasa de matriz-9 (MMP-9) y el factor de necrosis tumoral alfa (TNF*)31. Explantes aórticos de ratones PPAR* / presentan una producción exacerbada de agentes proinflamatorios como interleucina (IL) 6 en respuesta a estímulos inflamatorios como, por ejemplo, la adición de lipopolisacárido (LPS); en las mismas condiciones, los esplenocitos de ratones PPAR* / incrementan de forma exagerada la producción de IL-6 e IL-1231,32; estos datos indican que en los ratones PPAR* wt, la actividad PPAR* actúa modulando la intensidad de los procesos inflamatorios. Resultados similares se han podido demostrar en la práctica clínica; así, por ejemplo, la administración de fenofibrato a pacientes con hiperlipemia moderada produce un descenso significativo en las concentraciones plasmáticas de IL-6, TNF-*, interferón gamma (INF-*), fibrinógeno y proteína C reactiva (PCR)32,33.
El mecanismo de activación transcripcional o transactivación es minoritario en el control de los procesos antiinflamatorios e inmunomoduladores mediado por los PPAR. Los dos casos mejor estudiados son el control del metabolismo del LTB4 por activación de PPAR*16, que ya hemos comentado previamente, y el incremento de la expresión del gen que codifica la proteína I*B por agonistas de PPAR*34, gen cuyo promotor presenta un típico elemento de respuesta a PPAR o PPRE. La I*B es una proteína que actúa inhibiendo la actividad transcripcional de un factor de transcripción, el factor nuclear kappa B (NF-*B), que participa en el control y mantenimiento de los procesos inflamatorios.
Son los mecanismos de represión transcripcional, o transrepresión, los que están directamente implicados en el control de los procesos inflamatorios e inmunitarios por PPAR. Existen varios mecanismos posibles: ninguno de ellos implica una interacción directa del receptor nuclear con el material génico celular, aunque sí la presencia de ligandos de PPAR:
Secuestro de proteínas coactivadoras, necesarias para la actividad transcripcional de factores proinflamatorios, por el complejo PPAR-RXR. La inhibición de la expresión de fibrinógeno por activadores PPAR* se debe a una falta de actividad del factor de transcripción C/EBP ß (CCAT/enhancer-binding protein). A su vez, este déficit transcripcional se debe a una reducción de un cofactor imprescindible para C/EBP ß, la proteína GRIP-1 (glucocorticoid receptor-Interacting protein-1), secuestrado por el complejo PPAR*-RXR activado35.
Los PPAR pueden reprimir la transcripción génica al interferir con los factores de transcripción proinflamatorios NF-*B, STAT (signal transducer and activator of transcription), NFAT (nuclear factor of activated T cells) y AP-1 (activator protein-1)32,36-38, probablemente mediante interacciones proteína-proteína que dan lugar a la formación de complejos inactivos. La inhibición puede ser bidireccional, dependiendo de las cantidades relativas de cada uno de los receptores, la presencia o la ausencia de activadores, etc.39,40.
Inhibición por el complejo PPAR-RXR de los procesos de fosforilación y activación de enzimas integrantes de la cascada de fosforilación mediada por MAPK41.
Características farmacocinéticas
Los 3 fibratos más utilizados actualmente poseen en el hombre características farmacocinéticas comunes42:
Tienen una buena biodisponibilidad oral, que oscila entre el 60% para el fenofibrato no micronizado (el 100% en la forma micronizada) y el 100% para el gemfibrozilo y el bezafibrato.
Presentan una unión a las proteínas plasmáticas superior al 95%.
Se metabolizan en mayor o menor proporción por glucuroconjugación hepática.
Se eliminan, ya sea en forma inalterada o como glucurónido, por vía renal.
Las principales diferencias radican en la vida media del fármaco en el organismo: mientras que la del bezafibrato y el gemfibrozilo oscila entre 1 y 3 h, la del fenofibrato suele ser entre 19 y 27 h, y en las enzimas metabólicas afectadas por fenofibrato y gemfibrozilo. Aunque ambos fármacos son glucuroconjugados, lo son por isoformas de glucuronidasas diferentes, con la peculiaridad de que las isoformas que glucuroconjugan el gemfibrozilo, UGT1A1 y UGT1A3, son también las que mayoritariamente glucuroconjugan las estatinas. Además, el gemfibrozilo es un potente inhibidor de los citocromos CYP2C9 y CYP2C8, que también participan activamente en la eliminación metabólica de las estatinas43. Estas características hacen que el gemfibrozilo sea el fibrato con mayor potencial de interacciones farmacocinéticas de trascendencia clínica, con otros fármacos, especialmente las estatinas.
Reacciones adversas e interacciones
El fenómeno de proliferación peroxisómica inducido por la activación de PPAR* en el hígado de rata y ratón se combina con la aparición de hipertrofia e hiperplasia hepática y, a largo plazo, hepatocarcinogénesis; de hecho, los proliferadores peroxisómicos se comportan como carcinógenos no genotóxicos44 en roedores. La activación del PPAR* produce 3 efectos que conllevan la aparición de carcinogénesis: incremento de la expresión de las enzimas constituyentes del sistema de betaoxidación peroxisómica de ácidos grasos, con los consiguientes incremento en la producción de H2O2 y daño oxidativo del ADN, inhibición de la apoptosis e incremento de la proliferación hepatocitaria45. Si se tiene en cuenta la amplia utilización de los fibratos hipolipemiantes en la terapéutica humana durante períodos prolongados y con dosis elevadas, es evidente la trascendencia sanitaria que representaría determinar de una forma clara si el fenómeno proliferativo y, por consiguiente, el riesgo carcinogénico se producen en el hombre. Esta cuestión cobró especial trascendencia tras los resultados del estudio WHO con clofibrato, en el que se evidenció un incremento de la mortalidad por causas no cardivasculares, particularmente por cáncer, en el grupo tratado46,47. La inclusión de ese estudio en un reciente metaanálisis de la mortalidad relacionada con el tratamiento hipolipemiante es la principal causa de que el grupo de los fibratos sea el único grupo de fármacos hipolipemiantes que parece incrementar significativamente la mortalidad por causas no cardiovasculares48.
Aunque la polémica dista mucho de estar acabada, los datos que existen hasta el momento indican que la especie humana parece ser resistente al fenómeno de proliferación peroxisómica. De los 5 trabajos publicados en los que se estudia este fenómeno en pacientes tratados con fibratos, únicamente en 2 se detecta un ligero incremento en el número o el volumen peroxisómico de las biopsias estudiadas, resultados cuya relevancia, dada la alta variabilidad interindividual, los propios investigadores cuestionan49. Igualmente, la inmensa mayoría de los estudios de proliferación realizados en cultivos primarios de hepatocitos humanos han proporcionado resultados negativos50. Paradójicamente, se ha podido identificar la presencia de PPAR* y RXR en los hepatocitos humanos51, así como PPRE presentes en las zonas promotoras de los genes humanos homólogos a aquellos que responden al fenómeno proliferativo en rata o ratón52; estudios de cotransfección utilizando construcciones quiméricas de PPAR* humano parecen indicar que éste es perfectamente funcional53. En estos últimos años ha aparecido una serie de datos experimentales que indican que en el hombre no se producen el fenómeno de proliferación peroxisómica y la hepatocarcinogénesis relacionada con éste. Estos datos pueden resumirse en los siguientes puntos: a) la expresión de la isoforma PPAR* en hígado humano oscila en un 1-10% de la encontrada en hígado de rata o ratón54; b) hasta un 40% de la proteína PPAR* presente en el hígado humano es una forma polimórfica sin actividad transcripcional55; c) la capacidad de unión a un PPRE específico de las formas de PPAR* activas en hígado humano es, en comparación con el PPAR* de rata, entre 3 y 4 veces menor56, y finalmente, d) datos obtenidos en nuestro laboratorio indican que la administración continua, durante 8 semanas, de fibratos a dosis terapéuticas no incrementa la expresión de la acil-CoA oxidasa, enzima marcadora de la proliferación peroxisómica, en biopsias obtenidas de pacientes tratados57. Dado que las dosis de ligandos de PPAR* necesarias para producir el fenómeno de proliferación peroxisómica son muy superiores a las necesarias para inducir otros efectos mediados por PPAR*58, esta característica explicaría la efectividad terapéutica de los fibratos en la especie humana.
De forma muchos más clara y documentada, los fibratos en su utilización clínica pueden inducir los siguientes efectos adversos59:
Eritema cutáneo.
Incremento variable en el riesgo de aparición de litiasis biliar. Este fenómeno es mucho más marcado con el clofibrato y sus derivados que con los fibratos de segunda generación.
Alteraciones hepáticas, en la mayoría de los casos asociada a un ligero aumento transitorio y reversible de las transaminasas plasmáticas.
Miositis y miotoxicidad que, en casos muy excepcionales, puede conducir a la aparición de rabdomiólisis. Este efecto adverso grave puede potenciarse en caso de asociación de un fibrato con otro fármaco potencialmente miotóxico, como la niacina, la ciclosporina y las estatinas hipolipemiantes (especialmente, como ya se ha indicado, combinadas con gemfibrozilo)60.
En cuanto a las interacciones con otros fármacos que pueden presentar trascendencia clínica, cabría destacar, además de la ya comentada con estatinas, el desplazamiento de la unión a las proteínas plasmáticas de los anticoagulantes cumarínicos, que puede potenciar su efecto anticoagulante61. Esta interacción, de carácter farmacocinético, puede agravarse por un proceso farmacodinámico, ya que los fibratos de segunda generación, como ya se ha indicado y quizá con la excepción del gemfibrozilo, disminuyen en mayor o menor grado el fibrinógeno plasmático, potenciando así el efecto de los anticoagulantes orales.
Utilización terapéutica. Ensayos clínicos
Como ya se ha indicado en la introducción, los fibratos se utilizan para el tratamiento de las dislipemias, especialmente las que cursan con elevadas concentraciones de triglicéridos y bajas concentraciones de cHDL. Las dislipemias son uno de los principales factores de riesgo modificables que condicionan la aparición y la progresión de la aterosclerosis y las enfermedades cardiovasculares asociadas. Hoy no existe duda sobre el papel determinante de la hipercolesterolemia (básicamente en forma de cLDL) como factor de riesgo cardiovascular, y son cada vez más sólidos los datos que relacionan la hipertrigliceridemia moderada y, sobre todo, la hipoalfalipoproteinemia asociada (concentración anormalmente baja de cHDL) con la aparición de enfermedad cardiovascular62. Por lo tanto, la pregunta capital no es ya si los fibratos modifican las concentraciones de lípidos plasmáticos, sino si su utilización terapéutica reduce la progresión de la arteriosclerosis y, por tanto, la incidencia de accidentes cardiovasculares.
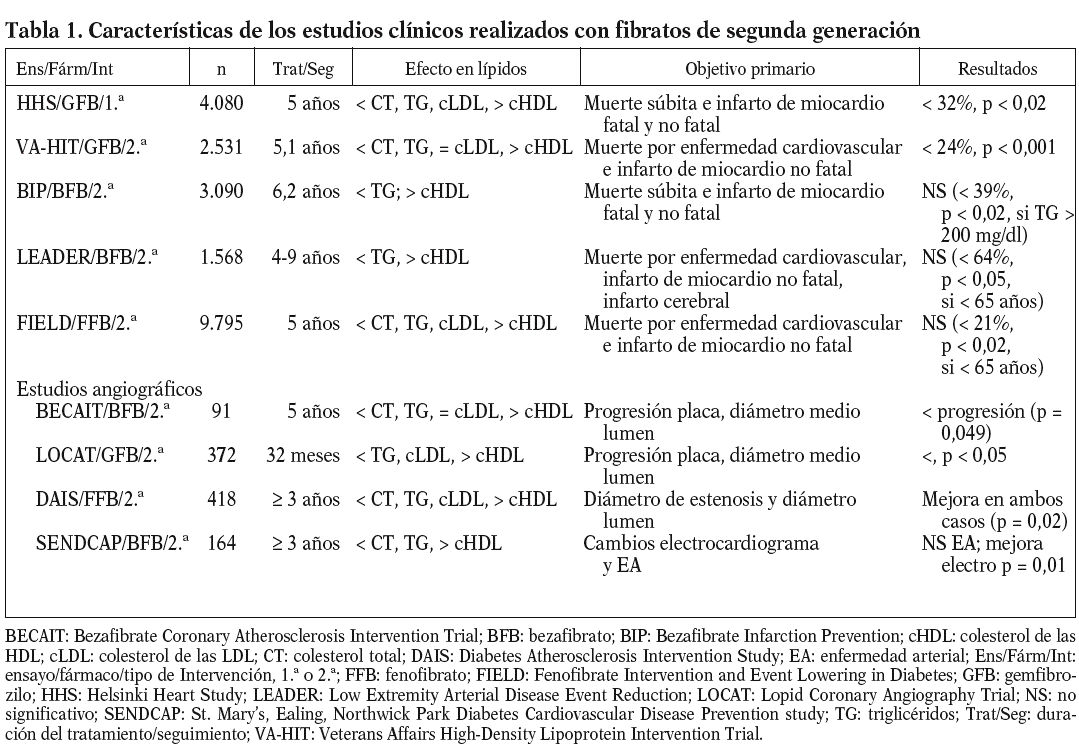
Hasta el momento se han realizado con fibratos 10 ensayos clínicos con determinación de accidente cardiovascular como objetivo final y 4 estudios angiográficos. De todos esos estudios, los 5 primeros en orden temporal, Newcasltle63, Edimburgo64, WHO46, CDP (Coronary Drug Project)65 y Stockholm Ischaemic Heart Disease66, fueron realizados con clofibrato. Estos estudios no serán comentados en la presente revisión por razones básicas: el clofibrato ya no se utiliza en la práctica clínica y, en segundo lugar, en estos estudios sólo se determinó el colesterol total como parámetro lipídico y no se incluyó a los pacientes con triglicéridos altos y cHDL bajo, que son los parámetros lipídicos en que el efecto de los fibratos es más intenso67.
En la tabla 1 se presentan las características más relevantes de los estudios clínicos realizados con fibratos de segunda generación67-69. Excepto en el caso del estudio HHS70, todos ellos pueden clasificarse como estudios de prevención secundaria. Salvo los estudios HHS y BIP71, en los que los pacientes tenían valores de colesterol elevados al incorporarse al estudio, la gran mayoría incorporó a pacientes con hipertrigliceridemia y/o bajas concentraciones de cHDL. Antes de la aparición del estudio FIELD72, la gran mayoría de los estudios indicaban un efecto positivo de la terapéutica con fibratos en la incidencia de accidentes cardiovasculares. Sin embargo, el análisis de todos estos estudios indicaba que el efecto beneficioso de los fibratos se centraba paticularmente en la reducción del riesgo cardiovascular en pacientes con diabetes tipo 2 o con características de síndrome metabólico como, por ejemplo, la dislipemia diabética68. Así, por ejemplo, en ensayos como HHS y VA-HIT26, la mayor reducción del riesgo vascular se obtenía en los pacientes con dislipemia diabética y obesidad (reducción del 78% en HHS) o diabéticos (reducción del 41% en VA-HIT). Incluso en los estudios con resultado general negativo, como el estudio BIP, análisis a posteriori indicaban una reducción del 39% de accidentes cardiovasculares en los pacientes con hipertrigliceridemia73. Sin embargo, tal como indica Vergès68, todos los indicios de un marcado efecto del tratamiento con fibratos en la prevención cardiovascular de diabéticos o pacientes con dislipemia diabética se obtenían a partir del análisis dción, el estudio angiográfico DAIS realizado en su totalidad con pacientes diabéticos74, no se diseñó con suficiente potencia como para demostrar una reducción de accidentes cardiovasculares tras la intervención con fenofibrato.
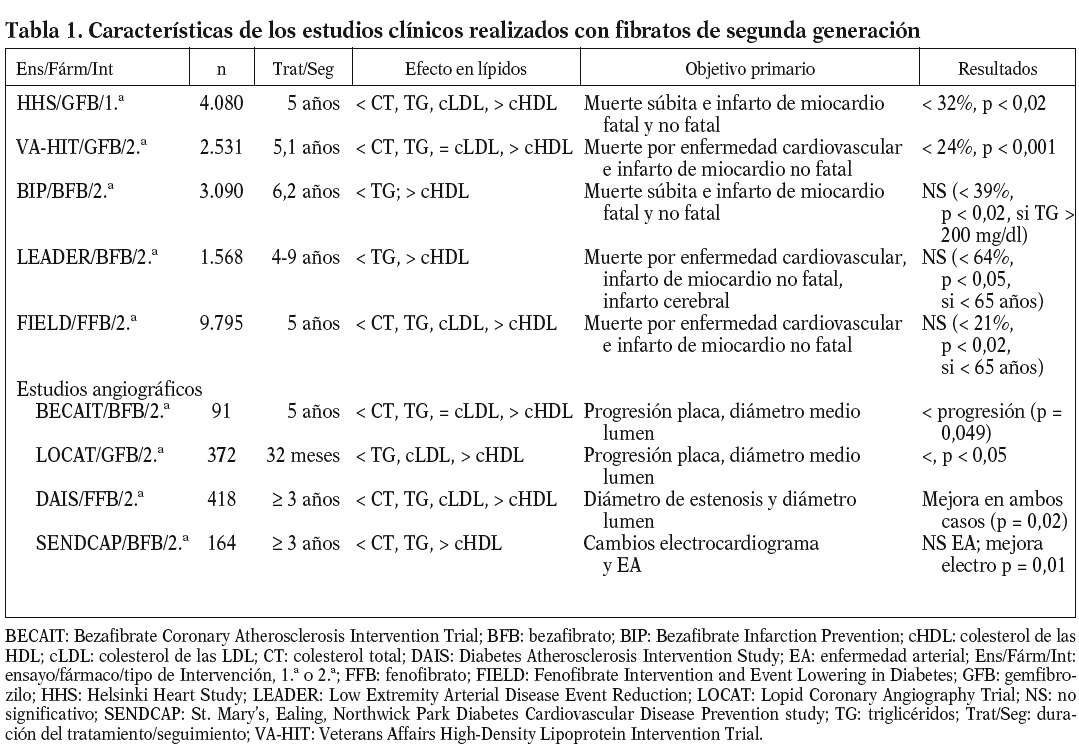
El estudio FIELD pretendía resolver esas dudas determinando el efecto del tratamiento con fenofibrato en los accidentes cardiovasculares en casi 10.000 pacientes diabéticos tipo 272. Por desgracia, los resultados obtenidos, publicados a finales del año pasado, han añadido más confusión al tema. Como se indica en la tabla 1, no se consiguió una reducción significativa del objetivo primario del estudio, la muerte por enfermedad cardiovascular e infarto de miocardio no fatal (el 11%; p = 0,16). Aunque se obtuvo una reducción significativa (p = 0,01) del 24% para el infarto de miocardio no fatal, ésta se acompañó de un incremento no significativo (p = 0,22) en la mortalidad cardiovascular. Los investigadores del estudio proponen algunas variables o características del estudio que podrían explicar los resultados obtenidos, en marcado contraste con lo esperado en función del análisis de los estudios previos al FIELD; entre estos factores podríamos citar la mayor proporción de pacientes tratados con estatinas en el grupo placebo, los cambios moderados o mínimos, aunque significativos, inducidos por el fenofibrato en los lípidos plasmáticos, sobre todo si se comparan con los de estudios de intervención previos, etc. Además, se podrían añadir 2 factores distorsionantes más de gran importancia:
1. En el estudio VA-HIT se obtuvo una marcada reducción del riesgo cardiovascular en presencia de unos cambios modestos en los lípidos plasmáticos (incremento en el cHDL del 6%); por el contrario, en el estudio BIP, no se obtuvo un efecto significativo en el objetivo primario del estudio (muerte súbita e infarto de miocardio fatal y no fatal), aunque se produjo un incremento del 14% en el cHDL. Podría ser que el efecto global de los fibratos sobre la enfermedad cardiovascular dependiera, en una mayor proporción, de los efectos "pleiotrópicos" antiinflamatorios que de sus efectos directos en las concentraciones de lípidos plasmáticos67. Si a este factor se le añade el hecho, como hemos comentado previamente, de que cada fibrato o ligando PPAR* puede presentar efectos particulares diferentes de los de los demás ligandos, dependiendo de su particular interacción con el receptor PPAR* y el reclutamiento de coactivadores, podríamos encontrarnos delante de una situación en la que el estudio FIELD deja en entredicho la eficacia del fenofibrato, pero no de los fibratos en su conjunto. Se tendrían que hacer estudios similares al FIELD con cada uno de los fibratos utilizados actualmente para solventar definitivamente este problema.
2. Estudios en modelos experimentales de envejecimiento en rata indican que la edad avanzada reduce la expresión y la actividad hepática del receptor PPAR*75; como consecuencia, los animales envejecidos presentan, en mayor o menor grado, una resistencia al efecto hipotrigliceridémico y al incremento en la expresión hepática de genes como L-CPT-I tras la administración de gemfibrozilo o bezafibrato76. Aunque evidentemente estos resultados no son directamente extrapolables a la especie humana, es interesante mencionar el hecho de que 2 de los 3 estudios clínicos con resultados negativos, LEADER77 y FIELD, incluyeron a pacientes con edades superiores a los 65 años. Análisis a posteriori en ambos estudios indican una marcada reducción de los accidentes cardiovasculares en la población de edad inferior a los 65 años, lo que podría indicar una ausencia de eficacia terapéutica de los fibratos en la población envejecida.
Es evidente, pues, que el estudio FIELD no ha dado portazo, ni mucho menos, a la investigación sobre el efecto beneficioso de los fibratos en la enfermedad cardiovascular. En cualquier caso, sin embargo, tal como el editorial que acompañó al estudio FIELD indica, los conocimientos actuales no preconizan un incremento en el uso del fenofibrato para el tratamiento de pacientes con diabetes ni para el de pacientes con adecuada concentración de cLDL78.