






La hiperlipemia familiar combinada (HFC) es la forma más prevalente de dislipidemia familiar con un origen multigénico y un patrón de herencia complejo. A este respecto, la HFC es un trastorno lipídico primario oligogénico debido a la interacción de variantes y mutaciones genéticas con factores ambientales. Los pacientes con HFC tienen un mayor riesgo de enfermedad cardiovascular y, con frecuencia, presentan otras alteraciones metabólicas asociadas. A pesar de su relevancia en la prevención cardiovascular, la HFC suele estar infradiagnosticada y, recurrentemente, infratratada. En la presente revisión, nos centraremos en los avances más recientes en la HFC, con el objeto de incrementar su conocimiento y, en definitiva, contribuir a mejorar su control clínico.
Familial combined hyperlipidaemia (FCH) is the most prevalent form of familial hyperlipidaemia with a multigenic origin and a complex pattern of inheritance. In this respect, FCH is an oligogenic primary lipid disorder due to interaction of genetic variants and mutations with environmental factors. Patients with FCH are at increased risk of cardiovascular disease and often have other associated metabolic conditions. Despite its relevance in cardiovascular prevention, FCH is frequently underdiagnosed and very often undertreated. In this review, emphasis is placed on the most recent advances in FCH, in order to increase its awareness and ultimately contribute to improving its clinical control.
De forma coetánea, Goldstein et al.1, Hazzard et al.2 y Kwiterovich et al.3 describieron en diferentes cohortes un patrón de dislipidemia caracterizado por la presencia de múltiples fenotipos lipoproteicos en familiares de sujetos jóvenes supervivientes de un infarto de miocardio, al que denominaron hiperlipemia familiar combinada (HFC). Esta hiperlipidemia se identificó por fluctuaciones en los lípidos plasmáticos y una presentación clínica heterogénea, pudiendo expresarse de forma alterna como una dislipidemia mixta, hipercolesterolemia o hipertrigliceridemia aisladas, en combinación con niveles elevados de apolipoproteína (apo) B. Además, suele coexistir con otras alteraciones metabólicas como la obesidad, la resistencia a la insulina, la diabetes mellitus tipo 2, la hipertensión arterial, el hígado graso no alcohólico y el síndrome metabólico4. Por tanto, existe unanimidad en otorgar a la HFC un alto riesgo de enfermedad cardiovascular (ECV) prematura5. Hay que resaltar, que la prevalencia de ECV es casi cinco veces mayor en los varones con HFC comparado con las mujeres afectas de mayor edad y la mayoría posmenopáusicas6.
Dada la compleja naturaleza de la enfermedad, las variadas definiciones clínicas y la falta de criterios diagnósticos específicos, la HFC permanece todavía infradiagnosticada y, en consecuencia, infratratada7, a pesar de tener un alto riesgo de ECV. A ello contribuye, sin lugar a duda, el bajo conocimiento de esta dislipidemia tanto en la atención primaria como en la especializada. A continuación, en la presente revisión, nos centraremos en los principales avances en el área de la HFC, con el objeto de contribuir a mejorar su control clínico.
Prevalencia y diagnósticoLa HFC es una de las dislipidemias más frecuentes, con una prevalencia estimada del 1 al 3% de la población general y de hasta un 20% en pacientes con cardiopatía isquémica precoz, pudiendo alcanzar hasta el 38% de los supervivientes de un infarto de miocardio antes de los 40 años1,8. Sin embargo, y atendiendo a los expertos, esta prevalencia requiere una evaluación actualizada9.
La HFC se transmite en las familias con un fenotipo lipídico mixto y variable; la penetrancia aumenta con la edad, el estilo de vida no saludable y los factores ambientales, hecho que explica que esta dislipidemia tienda a desarrollarse a partir de la segunda década de la vida. La variación en el fenotipo tanto inter como intraindividual hace que su diagnóstico sea un reto. Además, la ausencia de un marcador genético o metabólico específico para el diagnóstico de certeza de la HFC justifica los diferentes criterios propuestos a lo largo de los años. Clásicamente, el fenotipo para establecer el diagnóstico de HFC consistía en la presencia de hipercolesterolemia o hipertrigliceridemia aisladas o dislipidemia mixta junto con antecedentes familiares de primer grado de ECV prematura, excluidas otras causas de dislipidemia1. En un importante estudio de seguimiento a cinco años, se constató que las concentraciones plasmáticas de colesterol y triglicéridos presentaban una variabilidad considerable a lo largo del tiempo en las familias con HFC. En cambio, las concentraciones elevadas de apo B-100 y una mayor proporción de partículas lipoproteicas de baja densidad (LDL) pequeñas y densas mostraron una mayor estabilidad en el tiempo y una consistente asociación con la HFC, incluso en los sujetos normolipidémicos10. En concordancia con estos hallazgos, se propuso un nomograma basado en la colesterolemia, trigliceridemia y concentración de apo B-100 para la predicción de la HFC11. El esquema diagnóstico más utilizado se expone en la tabla 1, y se establece con la presencia de dos criterios de inclusión y ninguno de exclusión12.
Criterios diagnósticos de la hiperlipemia familiar combinada
| A.- Criterios mayores de inclusión |
| CT ≥ 240 mg/dL o cLDL ≥ 160 mg/dL o TG ≥ 200 mg/dL con apo B > 120 mg/dL. Para los menores de 20 años, CT o TG por encima del percentil 90 según edad y sexo. |
| Hiperlipidemia múltiple en familiares de primer grado. |
| Afectación de aproximadamente el 50% de los familiares adultos de primer grado. |
| ECV prematura en familiares de primer grado (< 55 años en varones y < 65 años en mujeres). |
| B.- Criterios de exclusión |
| Xantomas en familiares de primer grado. |
| Hipercolesterolemia pura en niños de la familia estudiada. |
| cLDL > 300 mg/dL en familiares de primer grado. |
| Presencia de una causa secundaria de hiperlipidemia en el paciente o en los familiares afectos. |
| Presencia del genotipo de apo E E2/E2 en sujetos afectos. |
apo: apolipoproteína; CT: colesterol total; cLDL: colesterol de las lipoproteínas de baja densidad; ECV: enfermedad cardiovascular; TG: triglicéridos.
En esta hiperlipidemia el término «familiar» hace referencia a la importancia clínica del árbol genealógico y no a un posible carácter monogénico de la enfermedad. La HFC es un trastorno poligénico que se asocia con polimorfismos en más de 40 genes que participan en el metabolismo lipoproteico9. La HFC implica a polimorfismos genéticos que predisponen tanto a la hipercolesterolemia como a la hipertrigliceridemia, y que están relacionados con la disfunción del tejido adiposo, el aumento del flujo de ácidos grasos libres (AGL) al hígado, el dismetabolismo hepático lipídico y la alteración del aclaramiento de las lipoproteínas ricas en triglicéridos (LRTG) y de las LDL.
En la actualidad, se han descrito nuevos genes en la HFC, incluido el de la proproteína convertasa subtilisina/kexina tipo 9 (PCSK9), que participa en la regulación de los niveles del receptor LDL13. Así, la secreción hepática y las concentraciones plasmáticas de PCSK9 están aumentadas en la HFC14 y contribuyen al catabolismo retardado de las partículas LDL15. Aunque de forma muy infrecuente, se han referido variantes monogénicas raras que pueden explicar, por sí solas, la alteración fenotípica en algunos pedigríes16. La HFC puede coexistir con la hipercolesterolemia familiar, habiendo defectos monogénicos en la vía del receptor LDL que se superponen con varios genes menores, pero que interactúan y afectan el metabolismo de las LTRG17. Sin embargo, dado que las mutaciones genéticas comunes no explican totalmente la variabilidad observada en los fenotipos lipoproteicos de la HFC, la aplicación de las técnicas de secuenciación de próxima generación podría facilitar la identificación de nuevas variantes genéticas poco comunes, pero con un efecto significativo en el perfil lipídico.
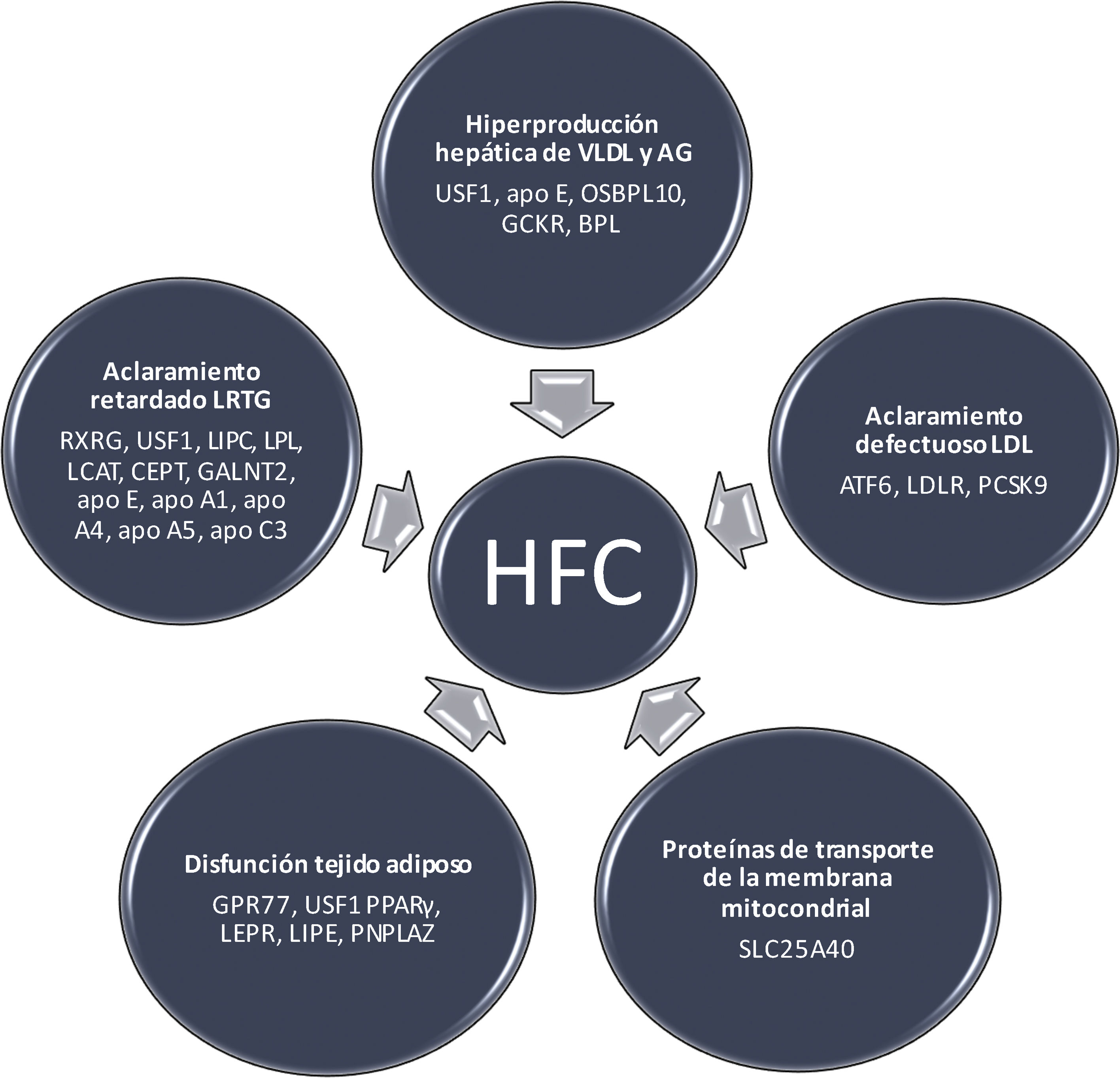
La base metabólica de la HFCSe han identificado diferentes mecanismos moleculares en la HFC (fig. 1), que se describen a continuación.

La característica principal de la HFC es la hiperproducción hepática de lipoproteínas de muy baja densidad (VLDL), con un incremento secundario de apo B. El aumento del flujo de AGL al hígado, la lipogénesis hepática de novo y los defectos de la β oxidación causan la acumulación de grasa hepática. En pacientes con diabetes, el incremento de la producción de VLDL aumenta cuando hay gran cantidad de grasa hepática y resistencia a la insulina. Varios genes involucrados en esta vía pueden desempeñar un papel en la patogenia de la HFC (fig. 1). En este sentido, el GCKR inhibe la glucoquinasa en el hígado y las células de los islotes pancreáticos, y algunas variantes en este locus se han asociado con lipogénesis de novo, β oxidación y niveles plasmáticos de triglicéridos18. Por su parte, el gen APOE, un ligando para el receptor LDL y apo E, puede conducir también a una sobreproducción de VLDL19. El gen OSBPL10, receptor intracelular de lípidos, está involucrado en la supresión de la lipogénesis hepática y la producción de VLDL; mutaciones en este gen pueden provocar un defecto en los procesos mencionados20. Finalmente, no debemos olvidar que el USF1 regula la transcripción de genes relacionados con el metabolismo de lípidos y glucosa21.
Aclaramiento retardado de las LRTGLas causas del retraso en el aclaramiento de las LRTG son complejas22. La lipoproteinlipasa (LPL) hidroliza las moléculas de triglicéridos transportados en las VLDL y los quilomicrones. Sin embargo, las partículas VLDL con menor contenido en triglicéridos son hidrolizadas por la triacil-glicerol lipasa hepática, codificada por el gen LIPC9. Utilizando una estrategia de genes candidatos, estudios de ligamiento y de asociación de todo el genoma (GWAS), se han identificado varios genes con un posible rol en las vías de eliminación de las LRTG; entre estos, se incluyen el LPL en el cromosoma 8p22, el gen LIPC en el cromosoma 15q21-23, apo CII y apo E en 19q13, y el grupo de genes apo CIII y APOA1/C3/A4/A5 en el cromosoma 11q23-2423 (fig. 1). La apo CIII impide la hidrólisis de los triglicéridos y el aclaramiento de las partículas remanentes, pudiendo ejercer efectos proaterogénicos. Este gen también se ha relacionado con la resistencia a la insulina y la diabetes tipo 2, frecuentes en la HFC. Otros genes que afectan la depuración de las LRTG incluyen el GALNT2 en el cromosoma 1q41-42, el LCAT en el cromosoma 16q22, el RXRG en el cromosoma 1q22-23, el USF1 en el cromosoma 1q22-23 y el gen CETP en el cromosoma 16q219 (fig. 1).
Tejido adiposo disfuncionanteEn los sujetos con HFC el recambio de triglicéridos en el tejido adiposo está reducido24. La lipólisis en el tejido adiposo implica la hidrólisis de los triglicéridos y en este proceso, la lipasa hormono-sensible (LHS), codificada por el gen LIPE, desarrolla un papel clave al estar incrementada su actividad en la HFC. En situación posprandial, esta enzima está inhibida por la insulina; al disminuir la acción insulínica en el ayuno y aumentar las hormonas contrainsulares, se produce el estímulo de la LHS del tejido adiposo y se liberan AGL que son captados por multitud de tejidos25. PNPLA2, que codifica la triglicérido-adiposo lipasa, es otro gen implicado en la lipólisis de los triglicéridos y en la disfunción del tejido adiposo26. Uno de los genes principales en la patogenia de HFC es el del factor de transcripción USF127. Las variantes genéticas en USF1 están asociadas con la lipólisis inducida por catecolaminas que está mediada por la fosforilación de los genes LHS y PNPLA2. En un estudio experimental, la inactivación del USF1 protegió contra la dislipidemia inducida por la dieta, la esteatosis hepática, la resistencia a la insulina, la obesidad y la aterosclerosis. El aumento del colesterol de las lipoproteínas de alta densidad (HDL) y la disminución de triglicéridos se acompañaron de un mayor gasto energético por la activación del tejido adiposo marrón28. Además, la inactivación de USF1 podría mejorar el aclaramiento de los triglicéridos plasmáticos. En ratones carentes de USF1 o con USF1 silenciado, se confirmó un efecto directo de este sobre la activación del tejido adiposo marrón después de una respuesta adrenérgica amplificada en adipocitos marrones y termogénesis aumentada inducida por noradrenalina28. Una variante en los genes del receptor 77 acoplado a la proteína G (GPR77) está relacionada con el almacenamiento de triglicéridos en los adipocitos; los individuos con esta mutación presentan menor almacenamiento de triglicéridos en los adipocitos y un perfil lipídico más aterogénico, indicando la posibilidad de que algunas formas de HFC puedan ser monogénicas16.
El receptor de la leptina, codificado por el gen LEPR, está generalmente producido por el tejido adiposo. Esta proteína participa en la regulación del metabolismo energético y del apetito. En este sentido, el polimorfismo Gln223Arg en el gen LEPR está vinculado con un mayor riesgo de HFC29.
Por su parte, el gen del receptor ɣ activado por el proliferador de peroxisomas (PPARG) regula la diferenciación de los adipocitos y la homeostasis de la glucosa. PPARG es, por tanto, otro gen involucrado en la disfunción del tejido adiposo30.
El tejido adiposo también secreta mediadores inflamatorios proaterogénicos entre los que se incluyen la resistina, adiponectina e interleucina-6 (IL-6)31. La resistina puede mejorar la expresión de PCSK9, con la consiguiente regulación a la baja de la expresión del receptor LDL. Sin embargo, deben intervenir otros mecanismos ya que el tratamiento con resistina en ratones desprovistos del gen PCSK9 reduce la expresión del receptor LDL32.
Los niveles de adiponectina se correlacionan negativamente con los de apo B y triglicéridos-VLDL, y se ha demostrado que la hipoadiponectinemia promueve un perfil lipídico aterogénico33. Por su parte, los niveles altos de IL-6 están relacionados causalmente con el riesgo de enfermedad cardiaca coronaria34.
El síndrome metabólico está presente hasta en un 65% de los sujetos con HFC, e incluso en mayor porcentaje si coexiste con ECV precoz35. Los primeros resultados que implicaron la resistencia a la insulina en la HFC fueron confirmados por estudios metodológicamente más complejos mediante el clamp hiperinsulinémico y demostraron una supresión deficiente de la insulina mediada por los niveles circulantes de AGL36. Hay que recordar que la liberación excesiva de AGL por los adipocitos altera la acción normal de la insulina al bloquear la oxidación de la glucosa y que la disponibilidad hepática de AGL da como resultado la secreción de apo B-100 y triglicéridos-VLDL37.
Defecto en el aclaramiento de partículas LDLLa alteración en el transporte por endocitosis de las LDL a las células puede conducir a HFC17. El ATF6 en 1q22-23, un sensor de la respuesta al estrés del retículo endoplasmático es otro gen que participa en el aclaramiento de las LDL en los sujetos con HFC. Por su parte, el gen PCSK9 está involucrado en la homeostasis del colesterol y relacionado con el fenotipo HFC13. También existe una interacción entre la proteína de unión del elemento regulador de esteroles 2 (SRBP2), la expresión de LDLR, la síntesis de colesterol y la expresión de PCSK9 en los hepatocitos.
Proteínas de transporte de la membrana mitocondrialLa secuenciación del exoma completo ha determinado un nuevo gen en un gran pedigrí de HFC. Este gen, SLC25A40, es un factor casual de hipertrigliceridemia y representa una nueva vía metabólica de gran interés como potencial diana terapéutica38 (fig. 1).
De forma complementaria, los GWAS y los estudios de ligamiento han apuntado otros genes que pueden contribuir a la patogenia de la HFC9.
TratamientoLas modificaciones del estilo de vida son la piedra angular del control de los pacientes con HFC. Los objetivos de presión arterial, control glucémico y el peso corporal son relevantes en el tratamiento de la HFC, por lo que remitimos al lector a los estándares de la Sociedad Española de Arteriosclerosis (SEA) 2019 para el control global del riesgo cardiovascular39.
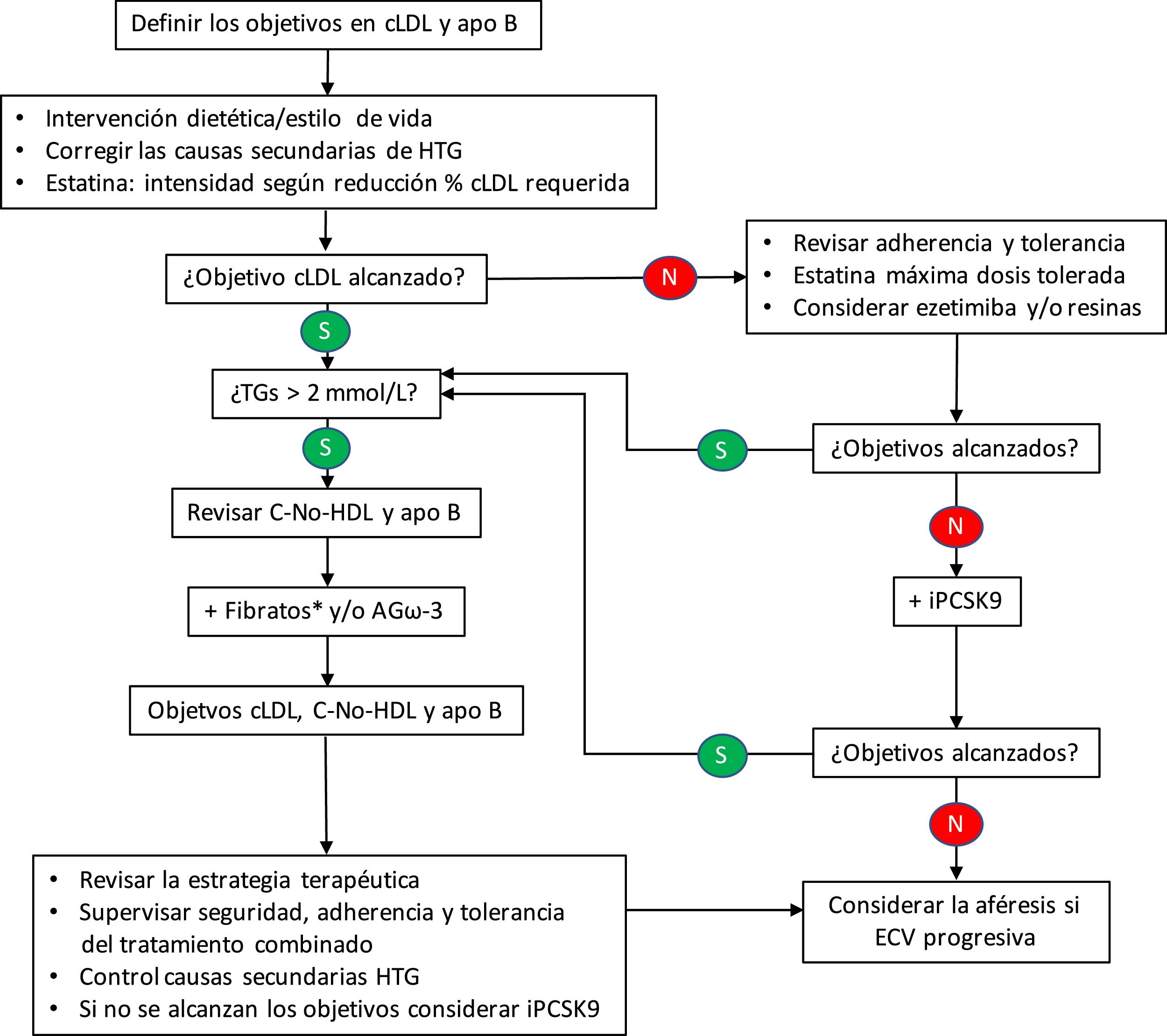
Por lo que se refiere al control lipídico, recientemente se ha publicado un algoritmo terapéutico para la HFC40, pero aún no se ha demostrado la rentabilidad y la aceptabilidad de este enfoque. En este algoritmo (fig. 2), la adición de un fármaco reductor de la trigliceridemia estaría indicada con una concentración > 2,0 mmol/L (175 mg/dL), siempre que se haya alcanzado el objetivo de colesterol LDL mediante el tratamiento con estatinas, con o sin ezetimiba. No debemos olvidar que la terapia combinada estatina-fibrato tiene un efecto beneficioso en todos los componentes del perfil lipídico41; si bien, es cierto que la respuesta puede depender de ciertos polimorfismos genéticos42.

Algoritmo terapéutico para la hiperlipemia familiar combinada.
apo: apolipoproteína; AGω-3: ácidos grasos omega-3; C: colesterol; cLDL: colesterol de las lipoproteínas de baja densidad; ECV: enfermedad cardiovascular; HDL: lipoproteínas de alta densidad; HTG: hipertrigliceridemia; iPCSK9: inhibidores de la proproteína convertasa subtilisina/kexina tipo 9; TGs: triglicéridos.
*Si el paciente está tratado con una estatina, el fenofibrato es el fibrato de elección al estar contraindicado el gemfibrocilo.
En la práctica totalidad de los esquemas propuestos, la hipertrigliceridemia es un factor determinante para las terapias adicionales dirigidas a la consecución de los objetivos de colesterol no HDL o de apo B. En este sentido, también se ha mostrado eficaz el empleo de los ácidos grasos ω-3 para modificar favorablemente el perfil lipídico, especialmente las LRTG y las distintas subclases de LDL43. Así mismo, se ha constatado el beneficio de la adición de nutracéuticos44, aunque, en este caso más orientado al control del número, tamaño y concentración de las partículas LDL.
FinanciaciónEste artículo ha sido financiado con una ayuda sin restricciones por Akcea Therapeutics. El patrocinador no ha intervenido en la elaboración ni el contenido del mismo, que expresa exclusivamente la opinión de los autores.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.