




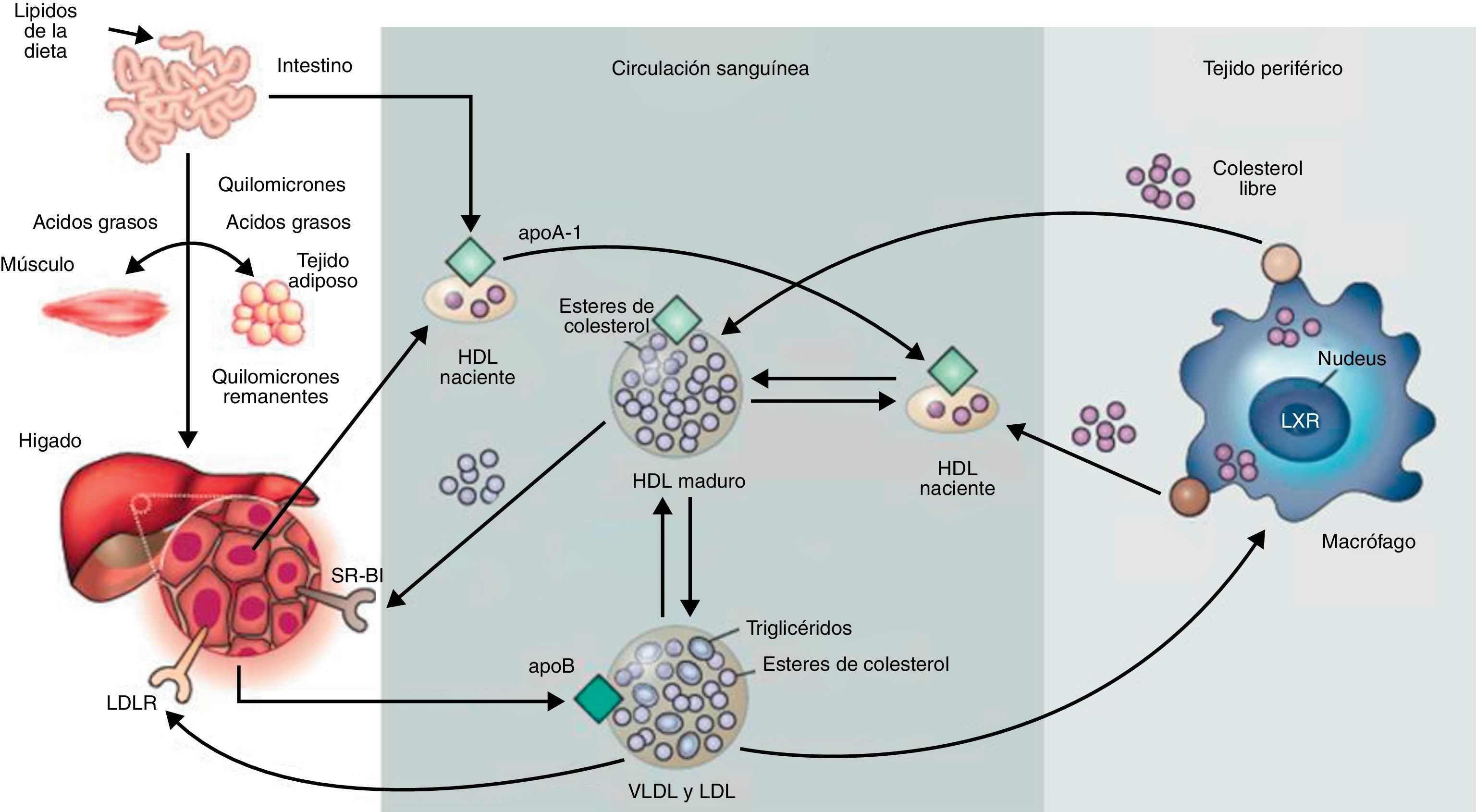
Las lipoproteínas son complejos macromoleculares solubles cuya función es el transporte de lípidos desde los tejidos de origen a los tejidos donde son almacenados o consumidos. Están formadas por un componente proteico (apolipoproteína) que, junto con los fosfolípidos y el colesterol libre, componen la cubierta de superficie que rodea al núcleo hidrofóbico compuesto por triglicéridos y ésteres de colesterol. Distintas combinaciones de lípidos y de proteínas dan a lugar a lipoproteínas de diferente densidad: quilomicrones, lipoproteínas de muy baja densidad (VLDL), lipoproteínas de baja densidad (LDL), lipoproteínas de densidad intermedia (IDL) y lipoproteínas de alta densidad (HDL). El componente proteico de las lipoproteínas, la apolipoproteína, permite la formación de la propia lipoproteína y, por tanto, el transporte de los lípidos (insolubles) por el riego sanguíneo. Además, tiene un papel fundamental en el metabolismo lipídico, puesto que es el componente implicado en la unión con los receptores celulares y en la activación de enzimas que actúan sobre las lipoproteínas. Se han descrito al menos 10apolipoproteínas en el plasma humano. Una de las clases más relevante en la práctica clínica, debido a sus características pro-aterogénicas1, es la apolipoproteínaB (apoB). Existen 2isoformas de apoB: la apoB-48 y la apoB-100. La apoB-48 es sintetizada en el intestino delgado. La apoB-100 es sintetizada exclusivamente en el hígado. Ambas formas están codificadas por el mismo gen APOB. La isoforma apoB-48 se genera como consecuencia de modificaciones post-transcripcionales que forman un codón de finalización prematuro. Por ello, la apoB-48 no presenta el extremo C-terminal de la apoB-100, necesario para la unión al receptor de LDL2. La apoB forma parte de varios tipos de lipoproteínas, tales como quilomicrones, VLDL, IDL y LDL. Los quilomicrones son sintetizados en las células epiteliales del intestino delgado, contienen apoB-48 y transportan los ácidos grasos de la dieta a los tejidos, donde son consumidos o almacenados. Los quilomicrones remanentes que han perdido gran parte de sus triglicéridos pero aún contienen colesterol son internalizados por el tejido hepático. En el hígado se empaquetan las VLDL, que contienen apoB-100 y que van cargadas de triglicéridos cuando la dieta es excesivamente rica en ácidos grasos. El exceso de hidratos de carbono en la dieta también se transforma en triglicéridos y se exportan en forma de VLDL. Las VLDL al llegar al músculo esquelético o al tejido adiposo activan enzimas con actividad lipasa sobre los triglicéridos, produciéndose la liberación de ácidos grasos. La pérdida de triglicéridos convierte las VLDL en VLDL remanentes, también llamadas IDL. Una posterior eliminación de triglicéridos forma las LDL. Estas LDL son muy ricas en ésteres de colesterol (alrededor del 37%) y contienen apoB-100 como la apolipoproteína principal3. Las LDL son, junto con las HDL, las lipoproteínas más abundantes en el plasma. Transportan colesterol en dossentidos: a los tejidos extrahepáticos o de vuelta al hígado, respectivamente. Las LDL son eliminadas de la circulación a través del receptor de membrana de LDL, capaz de reconocer la apoB-1004. Cuando una célula requiere colesterol, sintetiza el receptor de LDL, el cual se inserta en la membrana, difunde en esta y se ancla en zonas de la membrana ricas en clatrina. Cabe destacar que si bien la apoB-100 también forma parte de las VLDL, su dominio de unión al receptor no se encuentra disponible. La conversión de VLDL a LDL expone el dominio de unión al receptor (Fig. 1).
 o por vías alternativas. Las HDL son generadas en el intestino y en el hígado vía secreción apoA-I. ApoA-I recluta colesterol de estos órganos formando las HDL nacientes. En los tejidos periféricos las HDL nacientes promueven el flujo de colesterol desde los tejidos al hígado.Adaptado de Rader y Daugherty3.")
Metabolismo de las lipoproteínas. Incluye el transporte de colesterol y triglicéridos por la circulación sanguínea. El intestino absorbe los lípidos procedentes de la dieta y los empaqueta en quilomicrones que son transportados a los tejidos periféricos a través de la sangre. En el músculo y en el tejido adiposo la actividad lipasa rompe los quilomicrones y los ácidos grasos entran en los tejidos. Los quilomicrones remanentes son internalizados en el hígado. El tejido hepático forma VLDL a partir de apoB y lípidos, las cuales en los tejidos periféricos sufren lipólisis por la lipoproteína lipasa para formar LDL. Estos son absorbidos por el hígado vía el receptor de LDL (LDLR) o por vías alternativas. Las HDL son generadas en el intestino y en el hígado vía secreción apoA-I. ApoA-I recluta colesterol de estos órganos formando las HDL nacientes. En los tejidos periféricos las HDL nacientes promueven el flujo de colesterol desde los tejidos al hígado.Adaptado de Rader y Daugherty3.
Las LDL transportan colesterol y ésteres de colesterol a la pared arterial. A nivel de la íntima arterial las LDL son retenidas por proteoglicanos (PG), lo que favorece la modificación de estas lipoproteínas principalmente por oxidación (oxLDL) y agregación (agLDL). Las LDL modificadas se captan por receptores no regulados por colesterol intracelular, induciendo la transformación de macrófagos y células vasculares musculares lisas (VSMC) en células con una alta carga intracelular de lípidos neutros o células espumosas. La retención y la acumulación intracelular del colesterol procedente de LDL son procesos clave en el desarrollo de la placa de ateroma. Las placas con un alto contenido lipídico son muy susceptibles a la rotura, provocando episodios cardiovasculares adversos al favorecer la evolución de aterosclerosis a trombosis.
Retención de lipoproteínas de baja densidad en la íntima arterialLa retención y la modificación de lipoproteínas con apoB en la matriz extracelular de la íntima arterial son episodios clave en el desarrollo de la placa de ateroma («hipótesis de la respuesta a la retención»)5,6. Los PG son considerados los principales responsables de la retención y la modificación de LDL en la íntima arterial. Cuantitativamente, los PG más abundantes de la matriz extracelular son los sulfatos de condroitina (CS-PG). Los CS-PG tienen una alta capacidad de unirse a la apoB-100 de las lipoproteínas mediante interacciones electrostáticas que se producen entre los grupos sulfato y carboxilo de los glucosaminoglicanos (GAG) (cargados negativamente) y los residuos de lisina y arginina de la apoB-100 (cargados positivamente). Por un lado, se ha descrito que los PG aislados de áreas propensas a desarrollar lesión aterosclerótica se unen a mayores cantidades de LDL7. Por otro lado, el tamaño de las partículas de LDL también afecta a la unión de la LDL a GAG. Las LDL más pequeñas y densas, consideradas como un marcador de «fenotipo de lipoproteínas aterogénicas», presentan una interacción más fuerte con los PG8. Esta situación se encuentra con frecuencia asociada a resistencia a la insulina, diabetes tipo2 y enfermedad cardíaca coronaria. Las VSMC producen la mayor parte de los PG de la íntima arterial. Uno de los PG más secretados por estas células es el versicano. La interacción entre el versicano y la LDL produce cambios estructurales en la partícula de LDL que da a lugar a la formación de agregados de LDL (agLDL), altamente presentes en las lesiones ateroscleróticas9,10. Además del papel de las CS-PG en la modificación de LDL y su captación exacerbada por las células, se ha descrito que los lípidos intracelulares a su vez condicionan la producción de PG. La cantidad de PG sintetizado por VSMC aumenta cuando las células son tratadas con LDL oxidada (oxLDL)11. Las VSMC también sintetizan heparán sulfato (HS-PG), que puede ser secretado o formar parte de la superficie celular. En contraste con los CS-PG, que desempeñan un papel importante en la retención de LDL y la modificación en la íntima arterial, los HS-PG puede actuar como receptores potenciales para las lipoproteínas aterogénicas o facilitar la captación de ligandos por un proceso llamado transferencia de ligando a receptores de lipoproteínas, como el low-density lipoprotein receptor-related protein (LRP1). Finalmente, las VSMC producen importantes cantidades de colágeno y elastina. En concreto, la elastina extraída de la íntima aterosclerótica tiene una mayor capacidad para unirse a LDL que la extraída de la íntima arterial sana. La elastina parece unirse a las LDL a través de interacciones hidrofóbicas con los lípidos de las LDL en lugar de a través de interacciones apoB-10012. Además, las células espumosas producen una elastina con propiedades físicas alteradas que favorecen su rotura13,14.
Receptores lipoproteicos involucrados en la captación no regulada de colesterol ligado a lipoproteínas de baja densidadLa captación de LDL modificadas por macrófagos y por VSMC da lugar a la formación de células espumosas en la pared vascular. Pese a que se ha descrito una larga lista de agentes y enzimas que causan modificaciones de LDL in vitro, las principales modificaciones observadas en la íntima arterial son las oxLDL y las agLDL. Las oxLDL pueden generarse por el efecto en las LDL de distintas enzimas tales como la fosfolipasaA2 y/o la lipoxigenasa endotelial o la mieloperoxidada secretada por las células inflamatorias en las lesiones ateroscleróticas15. Los agLDL podrían ser generados por la degradación proteolítica de apoB-100 por la fosfolipasaA2, la fosfolipasaC o esfingomielinasa16, aunque la interacción directa entre PG y LDL, sin enzimas proteolíticas, causa agregación y fusión de LDL9. Existen 2grandes familias de receptores de lipoproteínas que participan en la captación de LDL por las células de la pared vascular: la familia del receptor de LDL y la familia de receptores scavenger.
Familia del receptor de lipoproteínas de baja densidadLos receptores que pertenecen a la familia LDLR son los siguientes: receptor de lipoproteínas de baja densidad (LDLR), receptor de lipoproteínas de muy baja densidad (VLDLR); LRP1, LRP1b; LRP2/megalina o glucoproteína 330 (gp330); LRP3 (se asemeja estrechamente a ST7); LRP4 (Corin); LRP5; LRP6; LRP8 (apolipoproteínaE del receptor2, apoER2); LRP10 y LRP11 (SorLA, LRP9). El LDLR fue el primer miembro de la familia descrito, y su función principal es la captación y la retirada del colesterol LDL del plasma. La función de estos receptores es tan importante que una expresión deficiente o mutaciones que dan lugar a fallos funcionales del LDLR causan una de las enfermedades con mayor riesgo de aterosclerosis prematura, la hipercolesterolemia familiar17. El VLDLR se expresa en las células endoteliales y musculares de la pared vascular y se ha sugerido que participa en la formación de células espumosas a partir de los macrófagos. También se ha descrito que este receptor, al ser regulado al alza por la hipoxia/isquemia, juega también un papel importante en la acumulación de lípidos en el miocardio durante la isquemia. La acumulación de lípidos mediante la sobreexpresión del VLDLR determina la supervivencia después de un infarto agudo de miocardio18.
El LRP1 es un receptor multifuncional que se compone de 2cadenas: la alfa extracelular y la beta anclada a la membrana, con una parte citoplasmática. Mediante su cadena alfa capta una gran variedad de ligandos y mediante la cadena beta participa en señalización intracelular19. En los seres humanos el LRP1 se encuentra sobreexpresado en lesiones ateroscleróticas avanzadas ricas en lípido20, y existen estudios clínicos que sugieren una relación entre las alteraciones en la expresión de LRP1 y la enfermedad coronaria21. Una de las características principales del LRP1 es que, a diferencia del LDLR, este receptor se regula al alza por el colesterol intracelular22. Se ha demostrado la regulación al alza de este receptor en VSMC expuestas a agLDL y en la pared vascular de cerdos hipercolesterolémicos mediante los factores de transcripción SREBP-2 (sterol regulatory element-binding proteins2)23. El LRP1 vascular también se regula al alza por la hipertensión24 y la hipoxia25, factores clave en la progresión de las lesiones. El LRP1 une agLDL, y ello provoca que las células vasculares se conviertan en células espumosas9, se transformen en células protrombóticas26 y alteren su migración27. En modelo animal de ratón, la inhibición del LRP1 en macrófagos aumenta la aterogénesis, revelando el papel de LRP1 en el reclutamiento de monocitos, en la regulación de la respuesta inflamatoria y en la actividad de metaloproteinasas28. También en modelo de ratón, la inhibición del LRP1, específicamente en células vasculares, da lugar a proliferación de VSMC, deterioro de la contractilidad vascular e hiperplasia intimal29. Todas estas alteraciones podrían explicarse, al menos en parte, por la función esencial de este receptor en señalización intracelular y sugieren que la inhibición de la expresión del LRP1 no es una buena estrategia para el control del desarrollo de aterosclerosis, al menos en el modelo de ratón. Estudios recientes han demostrado además que existe una estrecha asociación entre los altos niveles de expresión del LRP1 en el miocardio de pacientes con cardiomiopatía isquémica y la acumulación intramiocárdica de colesterol esterificado30. Como mecanismos subyacentes está la sobreexpresión de LRP1 en cardiomiocitos en situación de hipoxia/isquemia y la función del LRP1 en la captación selectiva de colesterol esterificado de las VLDL31. La acumulación miocárdica de lípidos neutros se relaciona con alteraciones en el metabolismo del calcio32. Estos datos sugieren que el receptor LRP1 es clave no solamente en la aterosclerosis, sino también en la función cardiaca.
El LRP2 es el mayor miembro de la familia, con aproximadamente 600kDa. Desde el punto de vista cardiovascular el LRP2, o glucoproteína 330, parece mediar la endocitosis y la degradación de la apoJ y de la apoM, apolipoproteínas antiaterogénicas que están presentes en las partículas de HDL, los quilomicrones, las VLDL y las LDL. El LRP2 también puede internalizar apoA-I y apoA-II, que son componentes estructurales de las HDL, lo que sugiere que el LRP2 contribuye a la regulación del metabolismo de las HDL33. Estudios de asociación genética han demostrado que las variaciones genéticas del LRP2 tienen influencia en el fenotipo de las lipoproteínas en pacientes con hipercolesterolemia34.
El LRP5 es un receptor que participa en la vía de señalización Wnt y juega un papel en la retirada de quilomicrones remanentes por los hepatocitos35. Recientemente se ha descrito que el LRP5 se regula al alza por lípido exclusivamente en células inflamatorias y que se colocaliza con macrófagos en lesiones ateroscleróticas36.
El LRP6 tiene funciones esenciales en el sistema cardiovascular porque los individuos que presentan la mutación R611C en este receptor tienen enfermedad coronaria difusa y enfermedad cerebrovascular, llegando a morir antes de los 50años. Esta mutación se asocia con altos niveles plasmáticos de colesterol, triglicéridos y glucosa, y también con presión arterial alta y prevalencia de diabetes37. Se ha demostrado que el LRP6 atenúa la señalización del PDGF, controlando la proliferación de las VSMC. Es por ello que la disfunción del LRP6 podría contribuir a una aterosclerosis acelerada en los individuos con la mutación R611C38.
El LRP8 se expresa en células vasculares, endoteliales y cardiomiocitos, y la mutación R9529 del LRP8 se asocia con enfermedad cardiovascular prematura e infarto agudo de miocardio39. Se ha encontrado una asociación entre algunas variantes del receptor LRP8 y presencia de partículas de LDL pequeñas y densas, con elevada afinidad por los PG y alta susceptibilidad de modificarse por oxidación.
Familia de receptores scavengerLa familia de receptores scavenger presenta al menos 8subclases diferentes de receptores de membrana y solubles (clases A, B, C, D, E, F, G y H) codificados por genes distintos y no relacionados. Los receptores scavenger reconocen una amplia variedad de ligandos, incluyendo oxLDL, células apoptóticas y patógenos40. Diferentes estudios en modelos animales de aterosclerosis han demostrado que el tipoA del receptor scavenger (SR-A) y el CD36 juegan un papel significativo en el desarrollo de células espumosas y en el desarrollo de la lesión aterosclerótica.
Los SR-A reconocen la porción oxidada de la apolipoproteína de la partícula de oxLDL y se expresan principalmente en los macrófagos pero también en VSMC y en células endoteliales41.
El CD36 reconoce los restos lipídicos de oxLDL y se expresa en monocitos, macrófagos, plaquetas, células endoteliales, adipocitos y VSMC42. Mientras que el receptor SR-A se regula al alza en respuesta a altos niveles de oxLDL, el factor estimulante de colonias de macrófagos (M-CSF) y ésteres de forbol, el receptor CD36 se sobreexpresa en respuesta a LDL y oxLDL, interleucina-4, M-CSF y ésteres de forbol. Por el contrario, los lipopolisacáridos y el TGF-β reducen los niveles de expresión del receptor CD36. Al igual que sucede con los receptores de la familia del receptor de LDL, la unión de ligandos a los receptores scavenger activa cascadas de señalización intracelular dando lugar a alteraciones funcionales tales como la apoptosis, la peroxidación lipídica o la diferenciación asociada a la formación de células espumosas. La activación de los receptores CD36 está asociada a la fosforilación y a la activación de MAP cinasas, lo que facilita la diferenciación de macrófagos a células espumosas. Se ha demostrado que la regulación al alza el CD36 por oxLDL está mediada por los factores de transcripción PPARγ (peroxisome proliferator-activated receptors) y que los cambios en la expresión génica de los macrófagos inducidos por oxLDL pueden atribuirse, al menos en parte, a la presencia de ligandos de PPARγ tales como HODE y 13-HODE en las oxLDL43. El efecto de los ligandos de PPARγ en la regulación al alza del CD36 podría compensarse por el efecto inhibitorio de los ligandos de PPARγ sobre el receptor SR-A44. La unión de ligandos a SR-A causa la activación de distintas vías de señalización relacionadas con proteína quinasa C y MAPK.
El LOX-1 (lectin-like oxidized low density lipoprotein receptor-1) se expresa en células endoteliales, macrófagos y VSMC, y también actúa como receptor de oxLDL. Se ha descrito que las células endoteliales estimuladas con factor de necrosis tumoral alfa (TNF-α) pueden secretar este receptor. Este receptor se ha encontrado en asociación con VSMC y macrófagos en lesiones ateroscleróticas avanzadas45.
Enfermedades metabólicas ligadas a la sobreproducción o a la falta de aclaramiento de lipoproteínas de baja densidad (tabla 1)Hipercolesterolemia familiar monogénica (mutaciones en el receptor de lipoproteínas de baja densidad)La hipercolesterolemia familiar (HF) es un trastorno genético autosómico dominante del metabolismo de lipoproteínas causado principalmente por mutaciones en la región codificante del gen del receptor de las LDL (LDLR). Su frecuencia estimada en su forma heterocigota es de 1:500. Los pacientes suelen presentar valores muy elevados de colesterol total y de colesterol LDL. Estos sujetos muestran rigidez arterial incrementada46, xantomas tendinosos y arco corneal. La característica clínica más importante de la HF es el alto riesgo de desarrollar enfermedad cardiovascular. Existe variabilidad en la concentración de LDL plasmática entre sujetos debido a factores como el tipo de mutación, otros genes del metabolismo lipídico, el sexo, la edad o la dieta. Los métodos recomendados para el diagnóstico de la HF se basan en el análisis del gen del LDLR y son altamente específicos. El diagnóstico y el tratamiento precoces son muy importantes para prevenir el desarrollo de la enfermedad cardiovascular47.
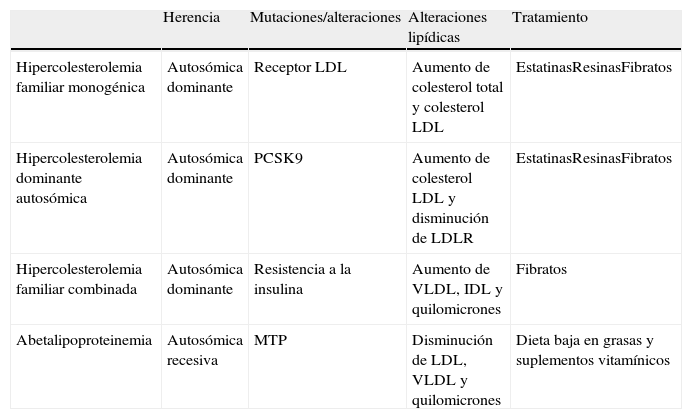
Principales patologías asociadas a la sobreproducción o deficiencia en el aclaramiento de lipoproteínas
| Herencia | Mutaciones/alteraciones | Alteraciones lipídicas | Tratamiento | |
| Hipercolesterolemia familiar monogénica | Autosómica dominante | Receptor LDL | Aumento de colesterol total y colesterol LDL | EstatinasResinasFibratos |
| Hipercolesterolemia dominante autosómica | Autosómica dominante | PCSK9 | Aumento de colesterol LDL y disminución de LDLR | EstatinasResinasFibratos |
| Hipercolesterolemia familiar combinada | Autosómica dominante | Resistencia a la insulina | Aumento de VLDL, IDL y quilomicrones | Fibratos |
| Abetalipoproteinemia | Autosómica recesiva | MTP | Disminución de LDL, VLDL y quilomicrones | Dieta baja en grasas y suplementos vitamínicos |
Existen otras hipercolesterolemias donde el defecto no está localizado en el gen del LDLR, sino en el PCSK9, el cual codifica una proteína de la familia de las subtilasas denominada NARC-1 (neural apoptosis-regulated convertasa1)48. Esta proteína interviene en la homeostasis del colesterol, favoreciendo el catabolismo del LDLR e impidiendo su reciclado a la superficie celular. Un incremento en la actividad de NARC-1 disminuye los niveles celulares del LDLR y aumenta las concentraciones plasmáticas de LDL, originando así un incremento en el riesgo cardiovascular. Por el contrario, las mutaciones que conducen a pérdida de función dan lugar a un aumento del número de LDLR, a menores concentraciones plasmáticas de LDL y, por tanto, a un menor riesgo cardiovascular49. Los pacientes con mutaciones de ganancia de función tienen niveles de LDLR incluso más bajos que aquellos con HF clásica. Sin embargo, el riesgo cardiovascular es más controlable, puesto que responden mejor al tratamiento con estatinas.
Diabetes y resistencia a la insulina. Hiperlipidemia familiar combinada (sobreproducción de lipoproteínas de muy baja densidad)La hiperlipidemia familiar combinada (HFC) es la hiperlipidemia más frecuente en pacientes supervivientes a infarto de miocardio antes de los 60años. Pese a que se ha determinado que presenta un carácter hereditario, se desconoce cuál es el defecto genético causante. La patogenia de la enfermedad se asocia con anormalidades en el metabolismo lipídico e insulinorresistencia en el tejido adiposo y muscular50. Las alteraciones del metabolismo de las lipoproteínas son 2-3veces más frecuentes en la población diabética que en la no diabética. Desde el punto de vista del riesgo cardiovascular, las concentraciones de lípidos en los pacientes con diabetes tipo1 tratados con insulina y en ausencia de nefropatía son similares a los de la población general. En cambio, en situación de mal control, la lipólisis favorecida por la insulinopenia condiciona un aumento de ácidos grasos libres y mayor producción hepática de VLDL. En situaciones de insulinorresistencia, la movilización de los ácidos grasos desde el tejido adiposo se incrementa, y el mayor flujo al hígado provoca una hiperproducción de partículas VLDL que se manifiesta en concentraciones elevadas de triglicéridos y apoB. Esta hiperproducción, unida al aclaramiento lento y disminuido de las partículas ricas en TG (quilomicrones, VLDL, IDL), condiciona un incremento de la concentración y del tiempo de permanencia en el plasma de estas partículas.
Abetalipoproteinemia (mutaciones en la proteína de transferencia de triglicéridos microsómica)La abetalipoproteinemia es un trastorno metabólico autosómico recesivo que se caracteriza por un defecto en el ensamblaje y la secreción de la apoB causada por mutaciones en la proteína MTP (microsomal transfer protein)51. Los triglicéridos de las VLDL provienen principalmente de la esterificación de los ácidos grasos de cadena larga en el hígado. Para el empaquetado hepático de los triglicéridos con apoB-100, ésteres de colesterol, fosfolípidos y vitaminaE se necesita la acción de la MTP. La mutación de la proteína provoca que el organismo sea incapaz de producir lipoproteínas LDL, VLDL y quilomicrones. Las principales manifestaciones del cuadro clínico son secundarias al defecto en la absorción y en el transporte de los lípidos, incluidas las vitaminas liposolubles52.