





Hypertriglyceridaemia has been associated with cardiovascular disease risk in humans for several decades. However, only recently, data from basic research, as well as from genetic and observational studies, have suggested triglyceride-rich lipoproteins (TRLs) as causal factors for atherosclerotic cardiovascular disease. Novel findings highlighting the relevance of TRL-derived lipolytic products (remnant lipoprotein particles “RLPs”), rather than plasma triglycerides or TRL themselves, as the true mediators in atherosclerosis, have contributed to explain a causal relationship through a number of direct and indirect mechanisms. Thus, experimental studies in animal models and in vitro cell culture methods reveal that RLPs, having sizes below 70–80nm, enter the arterial wall and accumulate within the sub-endothelial space. They then become involved in the cholesterol deposition of cholesterol in the intima in addition to several pro-inflammatory and pro-apoptotic pathways. In this review, a summary is presented of current understanding of the pathophysiological mechanisms by which TRLs and their lipolytic derived RLP induce the formation and progression of atherosclerotic lesions, and actively contribute to cardiovascular disease.
La hipertrigliceridemia se ha asociado durante décadas al riesgo de enfermedad cardiovascular en humanos. Sin embargo, ha sido recientemente, a partir de datos en investigación básica y de estudios genéticos y observacionales, que se ha sugerido que las lipoproteínas ricas en triglicéridos (TRL) son un factor causal en el desarrollo de la enfermedad cardiovascular. Nuevas evidencias sugieren que los productos derivados de la lipólisis de las TRL (remanentes lipoproteicos [RLP]), más que los propios triglicéridos o las TRL, son los mediadores moleculares en aterosclerosis, lo que ha contribuido a explicar mejor los mecanismos involucrados de forma directa o indirecta en la relación causal. Estudios experimentales, en modelos animales y en cultivos celulares in vitro, revelan que los RLP, con tamaños inferiores a 70-80nm, penetran en la pared arterial y se acumulan en el espacio subendotelial, contribuyendo activamente al acúmulo de colesterol, así como en procesos proinflamatorios y proapotóticos. Esta revisión recoge información sobre el conocimiento actual de los mecanismos fisiopatológicos por los que las TRL y sus derivados lipolíticos (RLP) inducen la formación y la progresión de lesiones ateroscleróticas y contribuyen de forma activa a la enfermedad cardiovascular.
Elevated level of plasma triglycerides (TGs), consequence of genetic disorders or derived from a variety of secondary causes (i.e. diabetes, alcohol intake, overweight), is a common lipid disorder that has been since decades associated to atherosclerotic cardiovascular disease (CVD).1 This association has acquired special interest over the recent years due to the disproportionate rise of overnutrition and obesity.2 So far, however, it is still unclear whether TGs have a direct causal role on atherosclerosis or just represent a CVD risk biomarker. Thus, the association between plasma TGs and atherosclerotic CVD has been confounded by its relationship to HDL3 and to metabolic disorders such as insulin resistance.4
Together with the cholesterol esters, the TGs are the most abundant circulating lipids. In blood, TGs are carried in the core of chylomicrons, very-low density lipoproteins (VLDL), and remnant lipoproteins (RLPs) generated during their metabolism including remnant-chylomicrons and remnant-VLDL, as well as intermediate-density lipoproteins (IDL) derived from VLDL.5 In contrast, cholesterol particularly concentrates in the low-density-lipoproteins (LDL) and high-density-lipoprotein (HDL) fractions despite being also present in the other types of lipoprotein fractions.
LDL-cholesterol remains as the primary treatment target for the management of dyslipidemias in both the primary and secondary CVD prevention settings.6–8 However, a substantial residual cardiovascular risk often remains in optimal treated patients,9 which has led to consider other therapeutic targets, including high levels of TG-rich lipoproteins (TRLs), in the prevention of atherosclerotic CVD.10,11 To this respect, Mendelian randomization and genome-wide association studies (GWAS) suggest a causal link between genetic variants associated to hypertriglyceridemia and risk of atherosclerotic CVD.5,12,13 This association is strongly supported by recent clinical studies. As such, Joshi et al.,14 using nuclear magnetic resonance have demonstrated risk-increasing associations of 13 TGs-subfractions with coronary heart disease, the strongest correlation being found with the TGs in the VLDL subfraction. In addition, the study by Suzuki et al.,15 which includes 1843 statin-treated subjects with clinical history of myocardial infarction (MI) drawn from the CHART-2 Study (Chronic Heart Failure Analysis and Registry in the Tohoku District), has evidenced that the TG level is a relevant part of residual risk for AMI recurrence in patients whose plasmatic LDL were well controlled (plasma level<100mg/dL). Also, different epidemiological studies have shown an independent association between circulating TGs and increased CVD-risk.3,6 This association, however, is attenuated when adjusted for variables such as HDL-cholesterol8 or the total concentration of Apo-B containing lipoproteins (non-HDL),6 which makes it difficult to establish a clear causal link between hypertriglyceridemia and CVD. In this respect, however, recent studies suggest that chylomicron- and VLDL-lipolysis products (RLPs) induce pro-atherogenic effects more effectively than TRLs themselves,16 which might contribute to explain the complex causal relationship between high TG-levels and atherosclerotic CVD.
This review aims to provide an overview of the most recent experimental and clinical evidence relating TRLs and their derivate RLPs with the composition of atherosclerotic lesions and plaque burden and the mechanisms by which these lipoproteins may actively contribute to the characteristics and evolution of the atherosclerotic lesions.
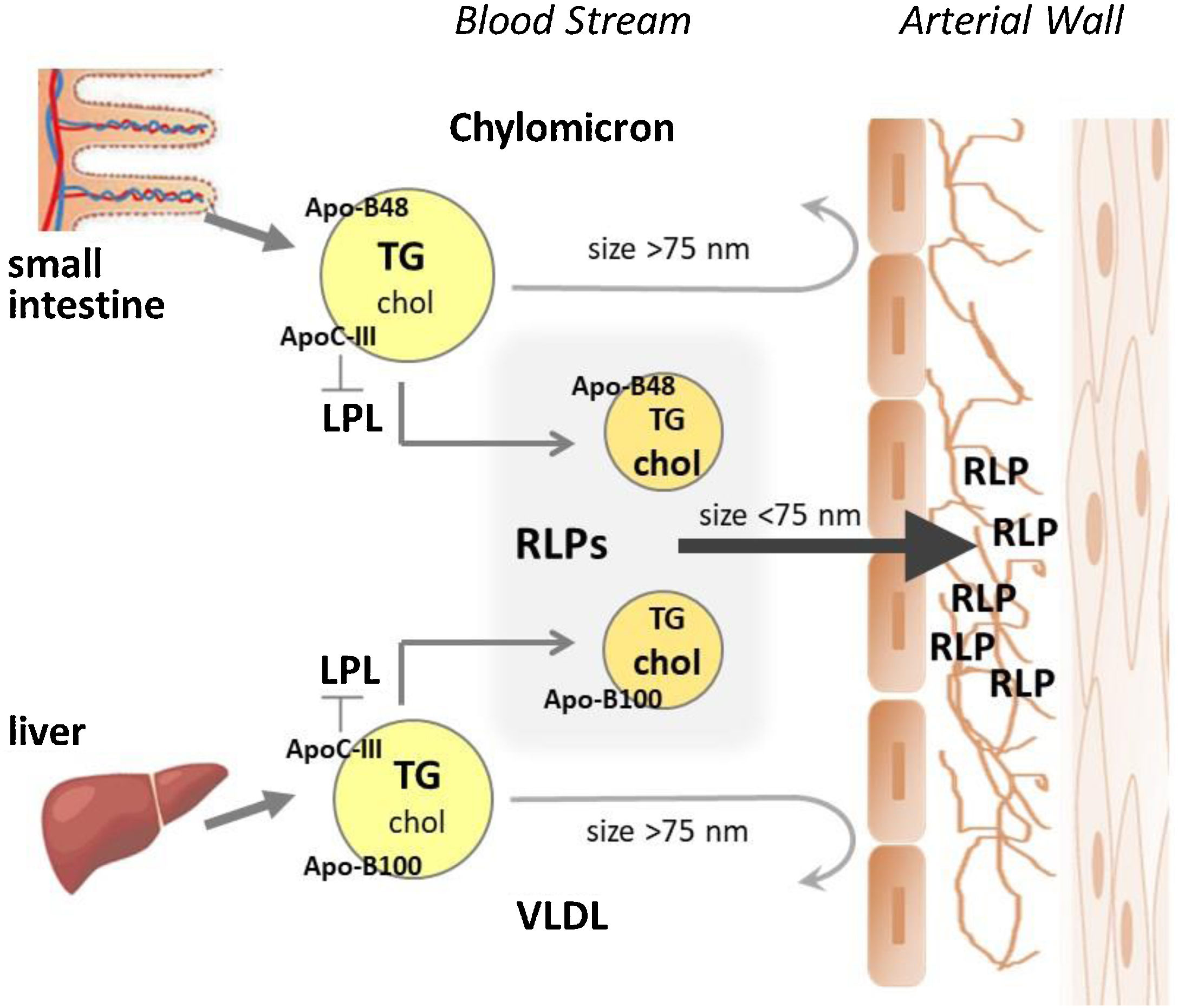
Triglyceride-rich lipoproteinsTG-rich lipoproteins (TRLs) are highly heterogeneous particles with a wide diversity in size, structure, and composition. Chylomicrons, the largest lipoprotein particles, are generated in the enterocytes upon dietary fat intake, whereas VLDL are synthesized and secreted by hepatocytes, being most of the TG transported by VLDL derived from glucose converted to TGs in the liver (Fig. 1). Through the chylomicrons and VLDL, TGs are transported to peripheral tissues, where they undergo hydrolysis and release fatty acids for energy production or storage. VLDL are present in plasma during unfasted and fasted states, unlike chylomicrons that mostly count for the postprandial concentrations of plasma TG in physiological states.
 synthesis and catabolism. TG: triglyceride; chol: cholesterol; RLP: remnant lipolysis products; VLDL: very-low-density-lipoproteins.")
Major lipid components of the TG-rich lipoproteins (TRLs) are diacylglycerols (DAG), triacylglycerols (TAG), cholesterol esters (SE), and phospholipids of the phosphatidylcholine class.17 In the clinic, cholesterol transported by the TRLs (non-LDL cholesterol and non-HDL cholesterol) is defined as remnant cholesterol, being suggested as the causal factor of the TRL-associated risk for ischemic heart disease, rather than TGs themselves.18 Thus, the study by Masson et al.,19 focusing in primary prevention patients without diabetes or lipid-lowering treatment, evidenced an independent association between calculated remnant cholesterol and the presence of subclinical carotid atherosclerotic plaques, assessed by 2D-mode ultrasound images. More recently, Lin et al.20 have also reported the association between circulating levels of remnant cholesterol levels and coronary atherosclerotic plaque burden assessed by computed tomography coronary angiography (CTCA) in 587 patients with suspected coronary artery disease (CAD), even in those with optimal LDLc levels.
As it occurs in the LDL, TRLs contain a copy of apolipoprotein (Apo) B, which refers to the full-length protein (ApoB100) in VLDL and IDL and to a splice variant containing the amino-terminal 48% of the full-length protein (ApoB48) in chylomicron-related particles. Thus, serum levels of ApoB48 reflects the number of chylomicrons and chylomicron-remnant particles.21 Postprandial ApoB48 levels have been shown to correlate with the presence of thin-cap fibroatheroma in nonculprit atherosclerotic lesions, as assessed by optical coherence tomography, and postprandial TRLs suggested as residual risk factor for CAD.22
TRLs also carried other several apolipoproteins including ApoE, ApoC-II and ApoC-III, in addition to a minor content in ApoD, ApoJ, ApoAIV, ApoAI, and serum amyloid A protein among other proteins.23 It is to notice that TRLs and RLPs are enriched in ApoE, a ligand of the low-density lipoproteins receptor (LDLR) family, specially of the LDL-related protein 1 receptor (LRP1), which is directly involved in the hepatic clearance of TRLs.24 LRP1 also mediates VLDL-TGs and CE uptake in extrahepatic organs, such as the heart, as demonstrated by Cal et al.,25 both in vitro and in vivo using a myocardial infarction experimental model in pig. Moreover, LRP1 plays a key role in the composition and evolution of atherosclerotic lesions,26–29 suggesting a LRP1-mediated role of TRL in the atherosclerotic process in the arterial wall.
TGs carried by the TRLs in the blood are rapidly hydrolysed by the lipoprotein lipase (LPL), through a process mediated by activation of ApoC-II and tidily modulated (LPL inhibition) by ApoC-III30,31 RLPs that generates free fatty acids and monoacylglycerols to be used by extrahepatic tissue cells for energy production or storage. As shown by in vitro studies, the ratio ApoC-III to ApoC-II is the main determinant for effective LPL inhibition,32 which is in line with current evidence of ApoC-III as risk marker of incident coronary artery disease.33
Similarly, ApoC-III plasma levels, measured by liquid chromatography/mass spectrometry, have been associated with increased risk for carotid artery plaque independently of the lipid pattern in the observational LIFE-Heart Study including 911 subjects with suspected stable coronary artery disease or myocardial infarction.34 In this respect, Kanter et al.,35 using an experimental mice model of type-1 diabetes mellitus have demonstrated that increasing levels of ApoC-III, consequence of insulin deficiency, result in elevated levels of TRLs and accelerated atherosclerosis, independently of variables such as HDLc, LDLc and glycated hemoglobin.
TRL lipolysis occurs at the luminal surface of the capillary endothelial cells through the complex between LPL and GPIHBP1.36 The latter is a GPI-anchored protein of capillary endothelial cells that has proved to be crucial for solving the LPL structure and stabilizing its activity,37,38 as well as for the margination of TRL along capillaries, facilitating LPL-mediated processing to occur.39
As consequence of LPL-mediated processing of TRLs, are generated remnant particles (RPLs) that are suggested as the triglyceride-rich lipoproteins with highest atherogenic capacity,40,41 due to their smaller size and therefore higher facility to enter the arterial intima, but also to their enrichment in CE content that occurs by action of the cholesteryl ester transfer protein (CETP). Indeed, RLPs are believed to be at least as atherogenic as LDL on a per particle basis.42 Thus, LPL-deficient mice, spite their high levels of TGs consequence of the lack of lipolysis and RLP-generation, exhibit a delayed formation atherosclerotic lesions of small size.43 In contrast, the apoE-deficient mice shows strong atherosclerosis, aligned with a plasma lipoprotein profile enriched in VLDL and their remnant lipolytic derivates.44 These findings appear to be consistent with the high prevalence of CAD in subjects with low RLPs removal capacity, as occur in familial dysbetalipoproteinemia (FD), a genetic dyslipidemia characterized by the accumulation in plasma of elevated levels of VLDL- and chylomicron- remnant particles.45
Presence of TRL and RLP in atherosclerotic lesionsAtherosclerosis is a complex pathophysiological phenomenon that involves lipid driven inflammation and oxidative stress processes resulting of the sustained and repeated exposure of the vascular endothelium and the arterial wall to systemic and local injury stimuli. Thus, high levels of lipids in the circulation and endothelial dysfunction are intimately connected, and entry and accumulation of Apo-B-containing lipoproteins in the vascular intima refer to early pathogenic events that promote atherosclerotic lesion formation.
Lipoproteins primarily flux into the arterial wall across the endothelial cells by transcytosis in specialized clathrin-coated vesicles of about 100nm in diameter.46 Therefore, lipoprotein size is a major limitation for entering the arterial intima through an intact endothelium, being the entry of particles bigger than 75nm completely blocked, which is the case of intact chylomicrons and large VLDL (also recognized as VLDL1). However, TRL-lipolytic components with sizes below this limit pass into the arterial wall and are retained in the subendothelial space (see Fig. 1), as demonstrated in genetically hyperlipidemic rabbits,47,48 contributing to subendothelial lipid accumulation. Supporting these findings, Nakano et al.,49 by using a HPLC (High Performance Liquid Chromatography) fractionation method and specific immunoassays described the presence of ApoB-48 carrying lipoproteins in extracts from human aortic atherosclerotic plaques obtained during autopsies for sudden cardiac death.
Retention of ApoB-rich particles in the intimal space is a key step in the atherosclerotic process and occurs by ionic interactions between positively charged residues of this apolipoprotein and the negatively charged components of proteoglycans in the extracellular matrix (ECM). TRL-remnants have lower affinity for proteoglycans than LDL through its ApoB unit.50 However, apolipoproteins such as ApoE and ApoC3 can also contribute to the binding of RLPs to the ECM-proteoglycan network.51,52
Using a perfusion experimental model in rabbit, Proctor et al.53 demonstrated that chylomicron-remnants and LDL permeate differently through the arterial tissue being the LDL located in deeper layers of the intima. Mass spectrometry technologies addressed to tissue imaging have made possible to gain detailed information on lipid composition and its spatial location in the atherosclerotic arterial intima. In this respect, Lethi et al.,54 by using time-of-flight secondary ion mass spectrometry (TOF-SIMS) have reported accumulation of CE, colocalizing with ApoB, near the intima-media border of human atherosclerotic lesions, whereas TGs accumulate almost exclusively at or near the luminal endothelium in all stages of atherogenesis with notably spatial segregation of cholesterol deposits, which evidences major differences in their capacity to move within the ECM network as suggested by Proctor et al.53 It is to notice, that TGs were also found in areas surrounding neovessels in the advanced lesions.54 Lipoprotein interaction with proteoglycans in the arterial intima also occurs indirectly through bridging molecules including LPL.55 LPL-dependent hydrolysis of TGs in the arterial intima has been considered to account for the location of TG-enriched particles and their potential effects for the course of the atherosclerotic process.54 In this respect, the use of TOF-SIMS has also led to identify the colocalization of LPL and monocyte chemoattractant protein (MCP)-1 with non-esterified fatty acids (NEFA)-rich areas in human atheroma plaque specimens, particularly from diabetic patients.56
Like LDL, TG-RLPs once entrapped in the subendothelial space are exposed to modifications (aggregation and fusion, enzymatic cleavage, and hydrolysis) and oxidative changes. In vitro studies have evidenced that proteolysis and hydrolysis of TG-rich particles induce their fusion and aggregation, being the formation of aggregates particularly produced after treatment with sphingomyelinase,57 lipolytic enzyme present in atherosclerotic lesions and directly implicated in atherogenesis.58
Modified TG-RLPs are internalized by macrophages and smooth muscle cells, turning into foam cells and consequently contributing to plaque formation and progression. Indeed, TG-RLPs are thought to be equal or even more atherogenic than LDL. Thus, remnant particles due to their larger size carry up to 40 times more cholesterol per particle than LDL59 and unlike LDL, TG-RLP are directly taken up by arterial macrophages and VSMC, without having to be modified,60 which strongly supports the concept of RLPs being much more potent at inducing formation of macrophage-and VSMC- derived foam cells in vivo than LDL on a per particle basis. In addition, LPL-generated NEFA carried in lipoproteins induced higher cytotoxic effects in cultured mouse peritoneal macrophages than those carried in albumin.61
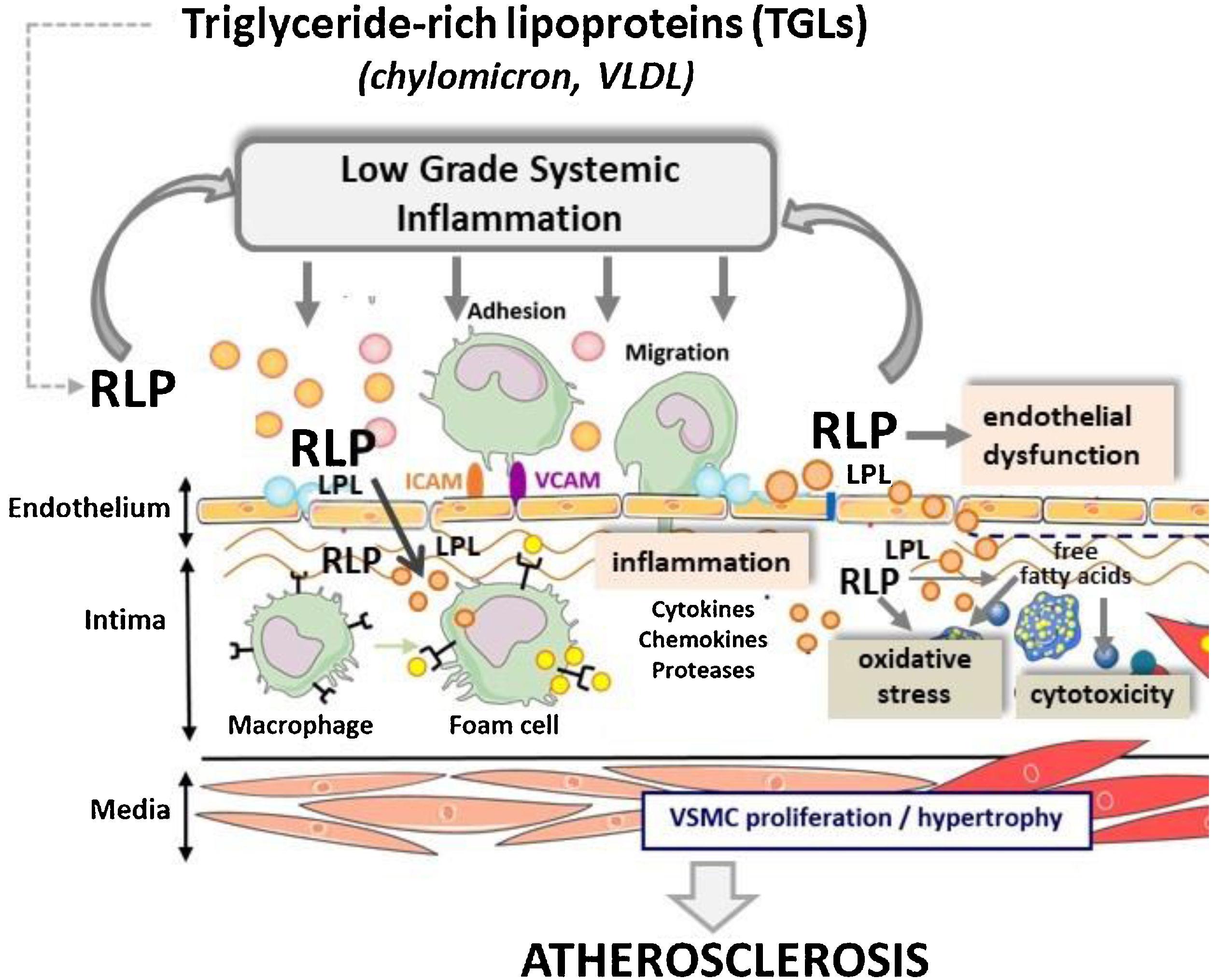
TRLs and RLPs induce endothelial dysfunction and inflammationAs discussed above, results from clinical and experimental studies implicate TRL and more specifically RLP in promoting atherosclerosis. Although the precise mechanisms involved remain still unclear, the current evidence links high concentrations of TRL-lipolysis products, resulting of LPL activity, along the blood- vascular endothelium interface with key pathological processes for atherosclerosis including endothelial dysfunction, oxidative stress, and inflammation (Fig. 2).
 and their remnant lipolytic products (RLPs) in the pathophysiology of atherosclerosis. VLDL: very low density lipoproteins; LPL: lipoprotein lipase.")
Endothelial dysfunction is systemic in nature, affecting vessels of different size throughout the arterial bed. Cardiovascular risk factors induce alterations in various physiologic functions of normal the endothelium including regulation of the permeability and vascular tone, and maintenance of anti-inflammatory and anti-thrombotic conditions.
TRL lipolysis products injured the vascular endothelium and increase the endothelial monolayer permeability due to the active rearrangement of junctional proteins, as demonstrated by in vitro studies in cultured human aortic endothelial cells. In addition, TRL induce disruption of proteins such as VE-cadherin and F-actin with the subsequent effect on the organization of the actin cytoskeleton and the cell viability.62 In this respect, the study by Lucero et al.63 focused on 40 patients with metabolic syndrome (MetS), has provided evidence that the TRL fraction from patients presenting endothelial dysfunction, measured by carotid-radial pulse wave velocity, had a TG over-enrichment when compared to TRL from patients with normal endothelial function. These authors further reported a positive correlation between TG content in TRL and the grade of inhibition of acetylcholine-mediated vasorelaxation of rat aorta rings.
TRL-lipolysis has shown to induce endothelial cell apoptosis dependent on TGF-β signaling, this accounts for the activation of the JNK/cJun pathway and induction of pro-inflammatory responses by TRL-lipolysis products in human aortic endothelial cells.62 In this respect, Aung et al.64 have evidenced a strong pro-inflammatory and pro-apoptotic response in cultured human aortic endothelial cells exposed to TRL lipolysis products and identified the activating transcription factor 3 (ATF3), a member of the CREB family, as a key regulatory gene involved in the apoptotic processes induced by TRL-lipolysis.
In addition, TRL-lipolysis products induce membrane lipid raft aggregation and activation of NADPH oxidase with production of reactive oxygen species (ROS), which may in turn increase endothelial permeability and promote leukocyte adhesion.65,66
InflammationInternalization of lipoproteins initiates the conversion of the healthy endothelium and vessel wall into an inflammatory niche, due to the increased expression of adhesion molecules. Thus, inflammatory response is considered a driving force for the progression of atherosclerotic lesions.
Blood monocytes are activated under pathological conditions such as hypertriglyceridemia, which facilitates their arterial recruitment.67 Interestingly, by Mendelian randomization analysis, elevated concentration of LDLc and remnant cholesterol were casually associated with ischemic heart disease. However only the later lipoprotein group was causally associated with systemic low-grade inflammation.68
Fatty acids, particularly the saturated spices are considered the most inflammatory components of the TG-rich lipoproteins.69 After their lipolytic release from lipoproteins, saturated fatty acids and oxidized phospholipids may induce inflammation by binding and activating various pattern recognition receptors, including Toll-like receptors of the innate immune system.70 Thus, Bleda et al.71 have shown that high levels VLDL and TGs provoke NLRP1 inflammasome gene activation in human aortic endothelial cell. Until now, however, the precise mechanisms underlying TRL-dependent inflammasome activation remain unknown.
Postprandial hypertriglyceridemia has also been linked to vascular inflammation and atherosclerosis. In this respect, lipolytic products from postprandial VLDL induced cytokine and integrin expression in peripheral blood mononuclear cells (PBMCs) and THP-1 monocytes that also showed increased adhesion capacity intracellular lipid droplet formation via activation of ERK2 and NFkB.72
TG- rich lipoproteins and RLP as therapeutic targets in atherosclerotic cardiovascular diseasePharmacological strategies for reducing plasma TG-levels have largely focused on (1) increasing TRL clearance by modulation of the peroxisome proliferator-activated receptor (PPAR) activity (e.g., by fibrates), and (2) reducing hepatic production of VLDL (e.g., by omega-3 fatty acids).
The ESC/EAS Guidelines for management of dyslipidemias published in 20196 recommend taking into account the use of triglyceride-lowering drugs in high atherosclerotic CVD risk patients with hypertriglyceridemia when lifestyle fail to lower triglyceride levels.
However, the relationship between changes in TG levels and reduction in CVD risk might not be straightforward. Thus, the recently multicentre placebo-control trial REDUCE-IT, which includes 8179 high risk patients, based established CVD or presence of CVD risk factors) has proved that Omega-3 Fatty acids are efficient in reducing cardiovascular death and non-fatal myocardial infarction in addition to other cardiovascular-ischemic events beyond statin action.73 However, a statistic association between reduction of events and triglyceride level reduction could not be proved arising consideration for involvement of non-lipoprotein mechanisms for the observed benefits.
Further large-scale clinical intervention trials are needed to better clarify whether lowering triglycerides and remnant cholesterol in those subjects with high concentrations can reduce the risk of atherosclerotic CVD. New therapeutic options, based on genetic evidence for reduced risk of CVD in families with genotypes involving markedly low triglyceride-level, are currently under development.
ConclusionsElevated plasma triglyceride levels are predictors of CVD risk in humans and high concentrations of RLPs derived from TRL lipolysis cause atherosclerotic CVD, in contrast to hypertriglyceridemia due to accumulation of large TRL that depicts low pro-atherogenic capacity in absence of TRL-lipolytic products. As summarized in Fig. 2, RLPs infiltrate the arterial intima and remain retained in the subendothelial extracellular matrix, where they may suffer further LPL-mediated lipolysis. Intimal RLPs are thought to be equally or more atherogenic than LDL. In this respect, differing from LDL, unmodified TRL-related particles are effectively taken up by macrophages and VSMC, which acquire a foam-cell phenotype, and RLP carry higher amount of cholesterol per particle than LDL. Moreover, due to LPL-induced lipolysis, RLPs directly contribute to accumulation of free fatty acids, which may account for the increased production of reactive species and the inflammatory background associated to pro-atherogenic TRL. Up to now, current therapies aimed to reduce cardiovascular risk though normalization of TRL-levels are far from optimal. New studies addressed to gain better understanding of the complex interplay between TRL metabolism and the whole spectrum of ApoB-containing lipoproteins might facilitate development of novel and more effective therapeutic agents for reduction of the residual CVD risk.
FundingThis work was supported by the Institute of Health Carlos III, ISCIII [FIS PI19/01687 to T.P., Red Terapia Celular TerCel- RD16/0011/0018 to L.B., CIBERCV to L.B.]; Spanish Ministry of Economy and Competitiveness of Science [PID2019-107160RB-I00 to L.B.] and cofounded by FEDER “Una Manera de Hacer Europa”. Secretaria d’Universitats i Recerca del Departament d’Empresa i Coneixement de la Generalitat de Catalunya [2017 SGR 1480]. We thank Fundación Jesus Serra and Fundación de Investigación Cardiovascular, Barcelona, for their continuous support. NMG is recipient of a “Beca Manuel de Oya” from FICYE, Spain.
Conflict of interestNone declared.
Nota al suplementoEste artículo forma parte del suplemento «Diagnóstico y tratamiento de las alteraciones del metabolismo de los triglicéridos: de la fisiopatología a la práctica clínica», que cuenta con el patrocinio de Akcea Therapeutics.