




The microsomal triglyceride transfer protein (MTP) is involved in hepatic and intestinal apoB secretion. We studied the effect of the functional MTP−493G/T polymorphism on fasting and postprandial lipoproteins in patients with familial combined hyperlipidemia (FCH) before and after treatment with atorvastatin.
MethodsEight FCH heterozygote carriers of the rare −493T allele were compared to 9 matched FCH homozygotes for the wild-type allele in a pilot study. Oral fat loading tests were carried out to measure triglycerides (TG) and apo B48 and B100 in the different fractions of triglyceride-rich lipoproteins (TRLs) before and after treatment with atorvastatin.
ResultsBefore treatment, TG were similar between the −493T allele carriers and non-carriers. In the T-allele carriers, a trend was observed for increased postprandial apo B48 and B100 concentrations in Sf >400 and Sf 60–400 compared to non-carriers. After treatment, fasting and postprandial TG were significantly lowered in carriers of the T allele, but atorvastatin had no effect on postprandial TG in non-carriers. Atorvastatin resulted in similar reductions of apo B48 and B100 in TRLs in both groups.
ConclusionThe MTP-493G/T polymorphism modulates postprandial apo B48 and apo B100 of TRLs in FCH. Atorvastatin decreases postprandial TG in T-allele carriers with FCH.
La Microsomal Triglyceride Transfer Protein (MTP) participa en la secreción hepática e intestinal de apoB. Hemos estudiado el efecto del polimorfismo funcional MTP−493G/T sobre las lipoproteínas en ayunas o posprandiales en pacientes con hiperlipidemia familiar combinada (FCH) antes y después de un tratamiento con atorvastatina.
MétodosOcho pacientes FCH heterocigotos para el alelo menos frecuente −493T fueron comparados con 9 pacientes FCH homocigotos para el genotipo salvaje. Tras una sobrecarga oral de grasa se cuantificaron los triglicéridos (TG) y las apoB48 y apoB100 de las diferentes fracciones ricas en TG, antes y después del tratamiento con atorvastatina.
ResultadosAntes del tratamiento, los TG eran comparables entre portadores y no portadores del alelo −493T. En portadores del alelo T, se observó una tendencia a mayores concentraciones de apoB48 y apoB100 en las fracciones Sf >400 y Sf 60–400, comparado con los no portadores. Tras el tratamiento, los TG basales y posprandiales fueron significativamente más bajos en portadores del alelo T, pero la atorvastatina no tuvo efecto sobre los TG posprandiales en pacientes no portadores de −493T. La atorvastatina indujo disminuciones en apoB48 y apoB100 de las fracciones ricas en TG tanto en portadores como en no portadores.
ConclusiónEl polimorfismo -493G/T de la MTP modula el contenido en apoB48 y apoB100 de las lipoproteínas ricas en TG posprandiales. La atorvastatina disminuye los TG posprandiales en pacientes FCH portadores del alelo T.
Familial combined hyperlipidemia (FCH) is a complex, polygenic disease characterized by increased VLDL production and decreased remnant clearance.1–6 Multiple genes have been associated with FCH, but none of them has been fully informative.2,7–11 The microsomal triglyceride transfer protein (MTP) has a key function in the intracellular lipidation and secretion of apolipoprotein (apo) B by the liver and intestine.12–14 MTP shuttles triglycerides (TG) from the smooth endoplasmic reticulum to the rough endoplasmic reticulum, where apo B is synthesized and mature TG-rich lipoproteins are produced. Mutations in the MTP gene lead to MTP deficiency associated with abetalipoproteinemia in homozygotes.12–14
Besides structural mutations leading to abetalipoproteinemia, the MTP gene seems polymorphic with multiple subtle changes in the MTP gene structure. The functional MTP−493G/T polymorphism has been associated with increased transcriptional activity with the T variant affecting TG metabolism.15,16 Increased postprandial generation of smaller, intestinally produced lipoproteins and reduced fasting TG plasma concentrations have been reported with the rare MTP−493T allele, but results were not always consistent.17–20 Furthermore, the MTP−493T polymorphism has been associated with decreased TG levels in untreated patients with heterozygous familial hypercholesterolemia.16,21 In addition, the MTP−493T polymorphism increased the TG lowering effect of atorvastatin in male subjects with heterozygous familial hypercholesterolemia.21 Moreover, homozygous carriers of the T-allele showed a stronger reduction of fasting TG after a diet low in saturated fatty acids and high in monounsaturated fatty acids and polyunsaturated fatty acids for a period of 3 months.22 The T allele has also been associated with increased insulin resistance and higher TG concentrations in males with the metabolic syndrome.23 Insulin resistance and elevated TG are characteristics of FCH and the MTP gene has been proposed as a candidate gene for the development of FCH. However, this could not be confirmed in a cohort of patients with the FCH phenotype based on fasting lipid profiles.24
This pilot study was initiated to evaluate whether the MTP−493G/T polymorphism is associated to postprandial TG, apo B48 and apo B100 changes in patients with FCH and whether the MTP−493G/T polymorphism influences the effects of atorvastatin on postprandial lipemia in FCH patients.
Material and methodsSubjectsThe Independent Ethics Committee of the University Medical Center Utrecht approved the study protocol and written informed consent was obtained from each participant. Seventeen unrelated FCH patients were recruited by genotype: participants were either heterozygous for the MTP−493T allele or homozygous for the MTP−493G allele. FCH was diagnosed with the following criteria: subjects were known with primary hyperlipidemia with varying phenotypic expression and they had all elevated plasma apoB concentrations (>1.20g/l) when untreated; at least one first degree relative had a different hyperlipidemic phenotype, and each index subject had a positive family history of premature cardiovascular disease disease defined as myocardial infarction or cerebrovascular disease before the age of 60. In addition, the patients fulfilled the following inclusion criteria: absence of xanthomas, and secondary factors associated to hyperlipidemia, body mass index (BMI) <30kg/m2, absence of apo E2/E2 genotype and no use of more than 3 units of alcohol per day.
Study design and treatment protocolAt inclusion, six out of seventeen FCH patients were using lipid-lowering drugs (atorvastatin 10mg, 40mg, 80mg, or simvastatin 20mg once daily). They stopped their medication 4 weeks before the first oral fat loading test was performed. After the first oral fat loading test the FCH patients were treated for 16 weeks with atorvastatin. Atorvastatin was chosen since statins are the first choice of drug treatment in FCH. Statins increase the fractional catabolic rate of both LDL and VLDL together with a slight reduction of hepatic VLDL secretion. The initial dose of atorvastatin was 10mg once daily. Every four weeks, the patients visited the outpatient clinic and fasting plasma lipids and apolipoproteins were measured. When plasma TG concentrations were above 2.00mmol/l and/or total cholesterol levels were above 6.5mmol/l, the atorvastatin dose was doubled, up to a maximum dose of 80mg after 12 weeks. After 16 weeks of atorvastatin, a second oral fat loading test was performed.
Oral fat loading testCream was used as a fat source; this is a 40% (w/v) fat emulsion with a P/S ratio of 0.06, which contains 0.001% (w/v) cholesterol, 2.8% (w/v) carbohydrates and 60g/l dextrose. After an overnight fasting period of 10hours, the subjects ingested cream (50g/m2) and were allowed to drink only water and sugar-free tea during the following 8hours. Peripheral blood samples were obtained in sodium EDTA (2mg/ml) before (t=0h), and at 1-hourly intervals up to 8h.
Analytical methods and laboratory techniquesBlood was placed on ice and centrifuged immediately for 15min at 800g at 4°C. After centrifugation, a protease inhibitor was added to the plasma as described.25 Plasma samples were stored at −20°C immediately after centrifugation. Apo E genotypes were determined as described earlier.26 MTP genotypes were determined as described by Karpe and co-workers.15,18 TG and total cholesterol were measured in duplicate by a commercial colorimetric assay (GPO-PAP and CHOD-PAP, Roche, respectively). HDL-cholesterol was determined as described earlier by Burstein using the phosphotungstate/MgCl2 method.27 Lipoproteins at time-points t=0, 2, 4, 6 and 8h were subfractionated by ultracentrifugation as described in detail.25,28
ApoB48 and apoB100 in Svedberg flotation (Sf) fractions representative for chylomicrons (Sf>400), VLDL1 (Sf 400–60) and VLDL2 (Sf 60–20) were quantitated by SDS-PAGE.25,28,29 Briefly, samples of each fraction were delipidated in a methanol/diethylether solvent system. After the first step, debris was removed with ice cold diethylether, and the proteins were dried by evaporation. The material was dissolved in sample buffer. Aliquots for apo B determination were stored at −80°C, and assayed within 3 months on 3–5% SDS-PAGE. The amount of apoB100 in the TRL fractions is usually too high to quantitate directly by SDS-PAGE; therefore, each sample was diluted 20 times with sample buffer and then loaded on the gel. For quantification of apo B48, each sample was loaded on the gel undiluted. The standard curve was made by delipidated LDL with known absolute amounts of proteins. In order to assess the equality of chromogenicities of apo B48 and apo B100, human chylous ascites, containing significant amounts of apoB48, was also delipidated and run on each gel.25 The proteins were stained with the Colloidal Blue Staining kit from Novex (Invitrogen, Carlsbad, CA, USA), containing Coommassie G-250, and destained by washing the gels at least four times with distilled water. After geometrical calibration, the gels were scanned with a B/W CCD camera type XC-77CE (Sony, Tokyo, Japan). For quantification of apo B48 and apo B100 a PC-based image analysis system was used (KS400 version 3.0 software, Carl Zeiss Vision, Oberkochen, Germany). Quantification of apo B48 and apo B100 was performed by a technician who was blinded to the code of the samples. The reproducibility of the method, calculated as 100% minus the coefficient of variation is 92.7%. The recovery of the apoB samples ranged from 70 to 80%. ApoAI was measured by nephelometry using apoAI polyclonal antibodies (OUED 14/15, Behring Diagnostics NV). The HOMA-IR index (=glucose (mmol/l)×insulin (mU/l)/22.5) was calculated.
Statistical methodsAll values in the text and tables are expressed as mean±standard deviation (SD). Mean±standard error of the mean (SEM) are used in the figures. Due to the small sample size, the groups were divided based on the presence of the T allele. Mean differences between carriers and non-carriers were calculated by the independent-samples t-test. Mean differences between untreated and treated FCH subjects were calculated by paired-samples t-test. TG were logarithmically transformed to obtain a normal distribution. Statistical calculations were performed using PASW 18.0 (IBM SPSS Inc. Chicago, IL, USA). Calculations for the area under the curves (AUC) were performed with GraphPad Prism version 5.0 (GraphPad Software Inc. San Diego, CA, USA). Statistical significance was reached when P<0.05 (two-tailed).
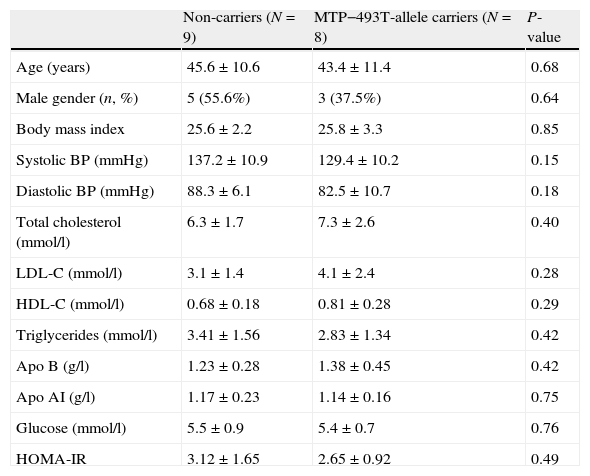
ResultsGeneral characteristicsA total of seventeen FCH patients were included. Nine patients were homozygote for the normal G allele, and 8 patients were heterozygote carriers of the MTP−493T allele. Baseline characteristics were similar between non-carriers and the T allele carriers: age (45.6±10.6 years vs 43.4±11.4 years), male gender (55.6% vs 37.5%), body mass index (25.6±2.2kg/m2 vs 25.8±3.3kg/m2) and HOMA-IR (3.07±1.87 vs 2.65±0.92). None of the patients expressed the apo E2/E2 genotype and the distribution of the apo E genotype was comparable between non-carriers and the T allele carriers. There were no significant differences in the variables shown in Table 1.
Baseline characteristics in familial combined hyperlipidemia patients in non-carriers and MTP−493T-allele carriers before treatment with atorvastatin. Data are given as mean±standard deviation unless stated otherwise.
| Non-carriers (N=9) | MTP−493T-allele carriers (N=8) | P-value | |
| Age (years) | 45.6±10.6 | 43.4±11.4 | 0.68 |
| Male gender (n, %) | 5 (55.6%) | 3 (37.5%) | 0.64 |
| Body mass index | 25.6±2.2 | 25.8±3.3 | 0.85 |
| Systolic BP (mmHg) | 137.2±10.9 | 129.4±10.2 | 0.15 |
| Diastolic BP (mmHg) | 88.3±6.1 | 82.5±10.7 | 0.18 |
| Total cholesterol (mmol/l) | 6.3±1.7 | 7.3±2.6 | 0.40 |
| LDL-C (mmol/l) | 3.1±1.4 | 4.1±2.4 | 0.28 |
| HDL-C (mmol/l) | 0.68±0.18 | 0.81±0.28 | 0.29 |
| Triglycerides (mmol/l) | 3.41±1.56 | 2.83±1.34 | 0.42 |
| Apo B (g/l) | 1.23±0.28 | 1.38±0.45 | 0.42 |
| Apo AI (g/l) | 1.17±0.23 | 1.14±0.16 | 0.75 |
| Glucose (mmol/l) | 5.5±0.9 | 5.4±0.7 | 0.76 |
| HOMA-IR | 3.12±1.65 | 2.65±0.92 | 0.49 |
Abbreviations: BP: blood pressure; apo: apolipoprotein.
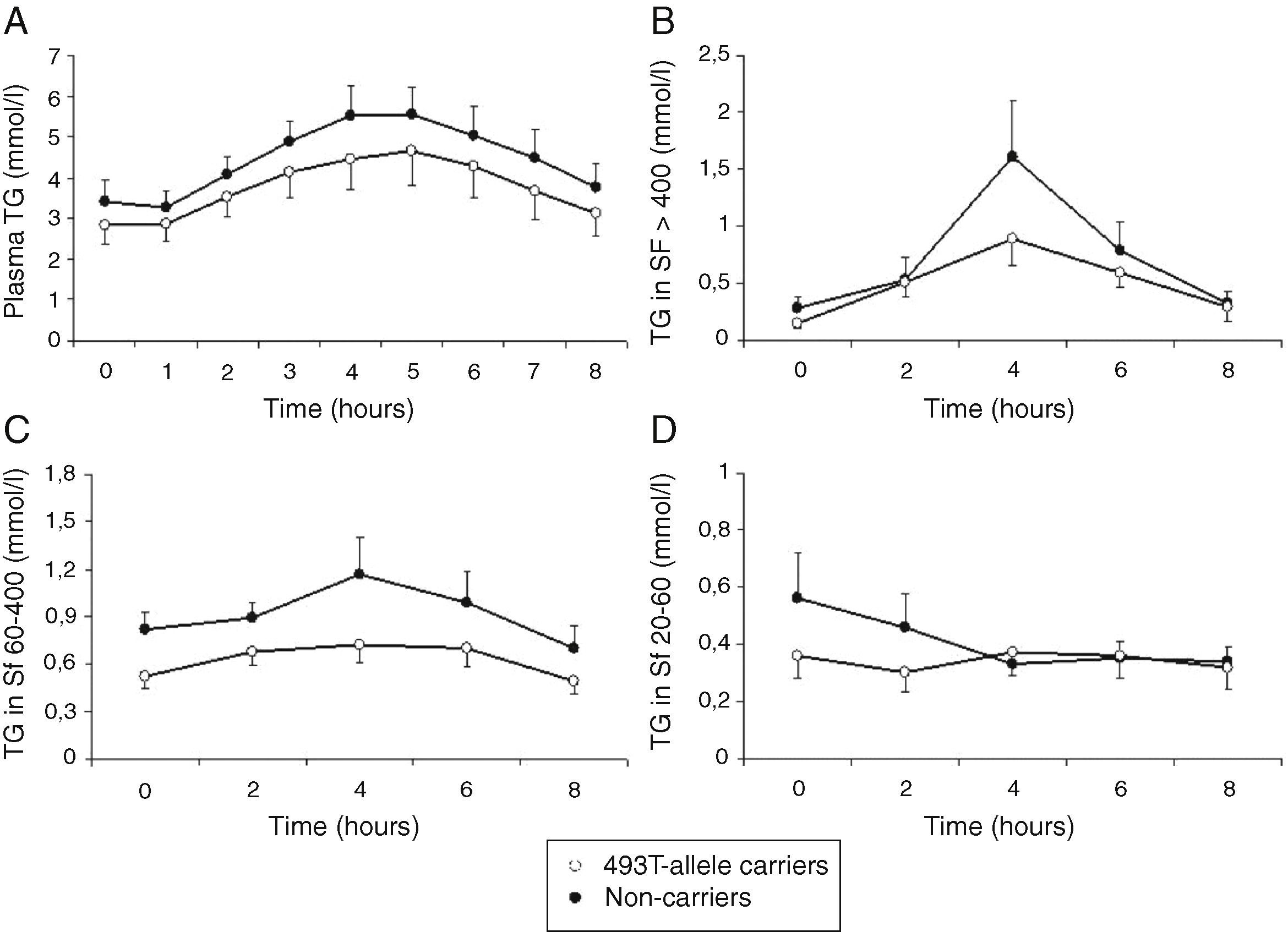
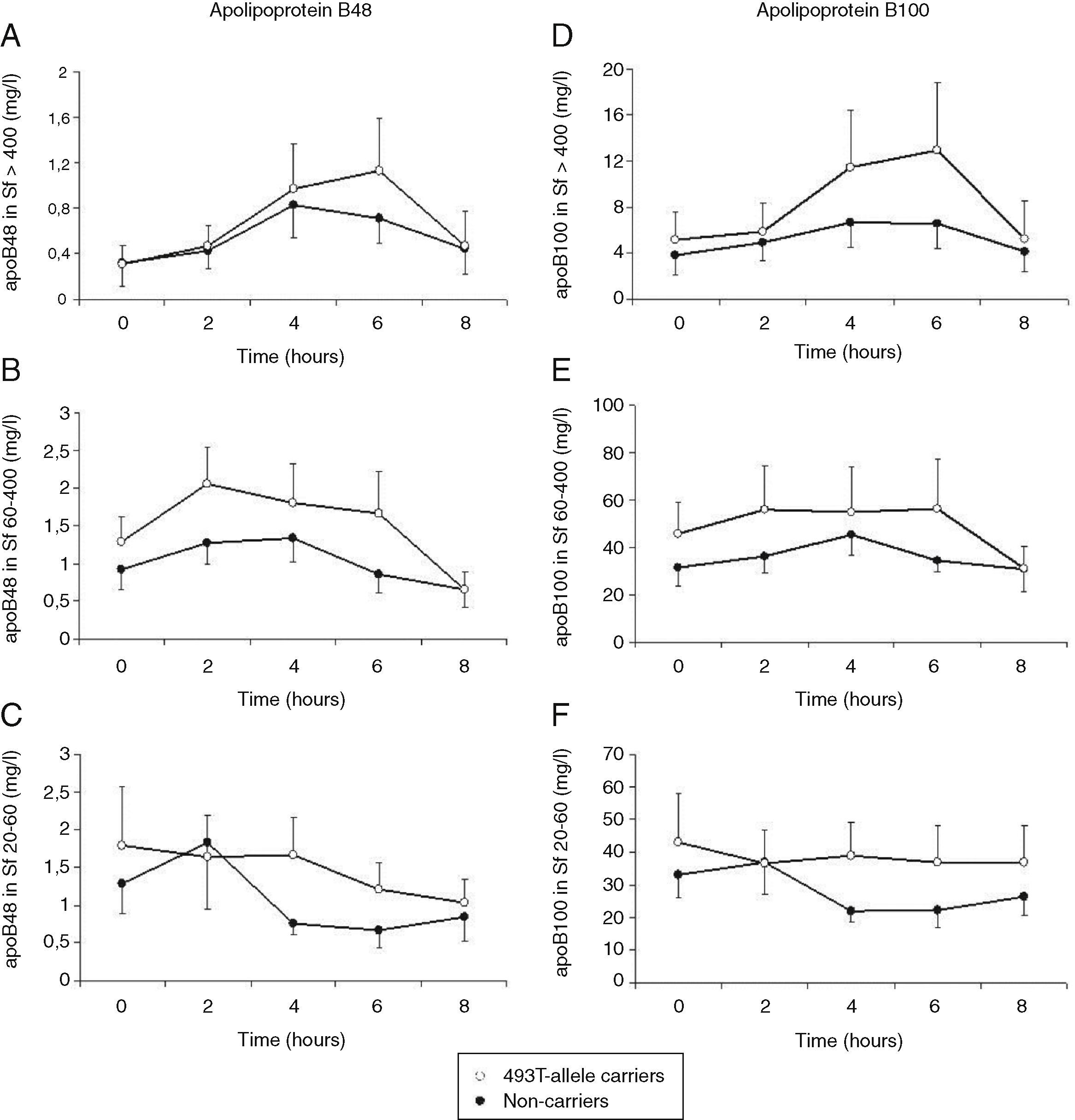
Total cholesterol remained unchanged in both groups postprandially. Postprandial triglyceridemia showed a similar pattern for the T allele carriers and non-carriers (Fig. 1A). In addition, no significant differences were found between the T allele carriers compared to the non-carriers in postprandial TG concentrations in the different TG-rich lipoprotein fractions (Fig. 1B–D). A non-significant trend for increased postprandial apo B48 and B100 concentrations in Sf >400 and Sf 60–400 in FCH carriers of the MTP−493T allele was observed (Fig. 2).
 (A), TG in Svedberg flotation fractions (Sf) >400 (B), 60–400 (C) and 20–60 (D) for untreated FCH patients in MTP−493T-allele carriers (open dots) and non-carriers (closed dots) during an oral fat load. No significant differences were found in plasma TG or any of the subfractions between −493T-allele carriers and non-carriers.")
Mean postprandial changes of plasma triglycerides (TG) (A), TG in Svedberg flotation fractions (Sf) >400 (B), 60–400 (C) and 20–60 (D) for untreated FCH patients in MTP−493T-allele carriers (open dots) and non-carriers (closed dots) during an oral fat load. No significant differences were found in plasma TG or any of the subfractions between −493T-allele carriers and non-carriers.
 B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated FCH patients in MTP−493T-allele carriers (open dots) and non-carriers (closed dots) during an oral fat load. No significant differences were found in apoB48 or apoB100 in any of the subfractions between −493T-allele carriers and non-carriers.")
Mean postprandial changes of apolipoprotein (apo) B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated FCH patients in MTP−493T-allele carriers (open dots) and non-carriers (closed dots) during an oral fat load. No significant differences were found in apoB48 or apoB100 in any of the subfractions between −493T-allele carriers and non-carriers.
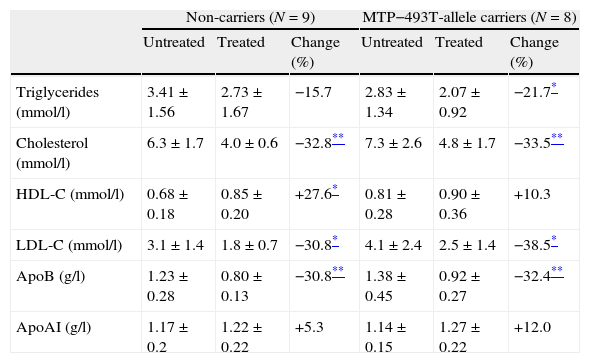
After 16 weeks, at the end of the titration period, 2 patients used 10mg, 5 patients used 20mg, 1 patient used 40mg and 9 patients used 80mg of atorvastatin. In the fasted state treatment with atorvastatin lowered plasma total cholesterol, LDL-C and apo B in the T allele carrier FCH patients as well as in the non-carriers (Table 2). Additionally, fasting plasma TG were significantly lowered by atorvastatin in the T-allele carriers, whereas HDL-C was increased by the non-carrier FCH patients after treatment with atorvastatin.
Changes in fasting lipid parameters before and after treatment with atorvastatin in familial combined hyperlipidemia patients in non-carriers and carriers of the MTP−493T-allele. Data are given as mean±standard deviation unless stated otherwise.
| Non-carriers (N=9) | MTP−493T-allele carriers (N=8) | |||||
| Untreated | Treated | Change (%) | Untreated | Treated | Change (%) | |
| Triglycerides (mmol/l) | 3.41±1.56 | 2.73±1.67 | −15.7 | 2.83±1.34 | 2.07±0.92 | −21.7* |
| Cholesterol (mmol/l) | 6.3±1.7 | 4.0±0.6 | −32.8** | 7.3±2.6 | 4.8±1.7 | −33.5** |
| HDL-C (mmol/l) | 0.68±0.18 | 0.85±0.20 | +27.6* | 0.81±0.28 | 0.90±0.36 | +10.3 |
| LDL-C (mmol/l) | 3.1±1.4 | 1.8±0.7 | −30.8* | 4.1±2.4 | 2.5±1.4 | −38.5* |
| ApoB (g/l) | 1.23±0.28 | 0.80±0.13 | −30.8** | 1.38±0.45 | 0.92±0.27 | −32.4** |
| ApoAI (g/l) | 1.17±0.2 | 1.22±0.22 | +5.3 | 1.14±0.15 | 1.27±0.22 | +12.0 |
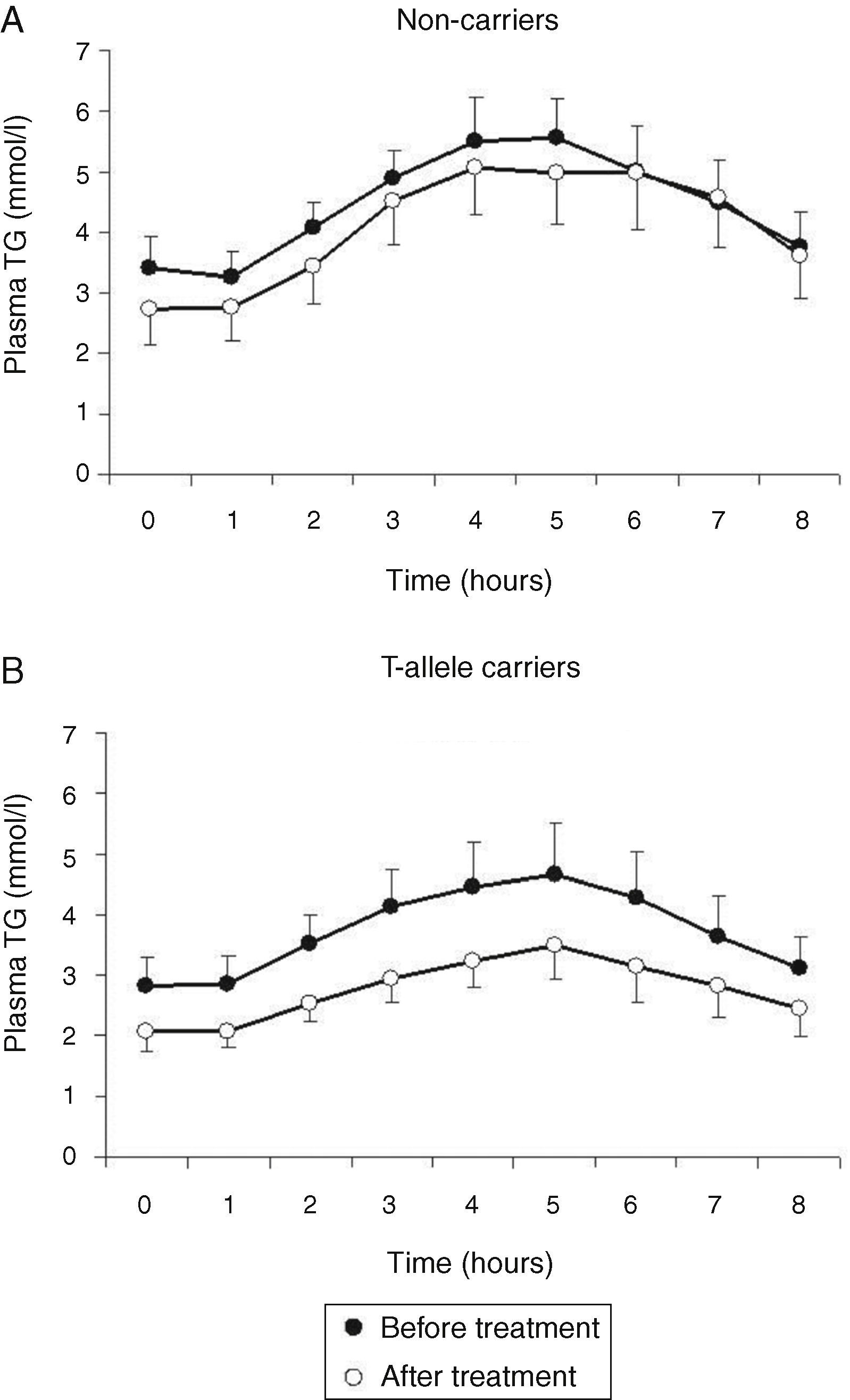
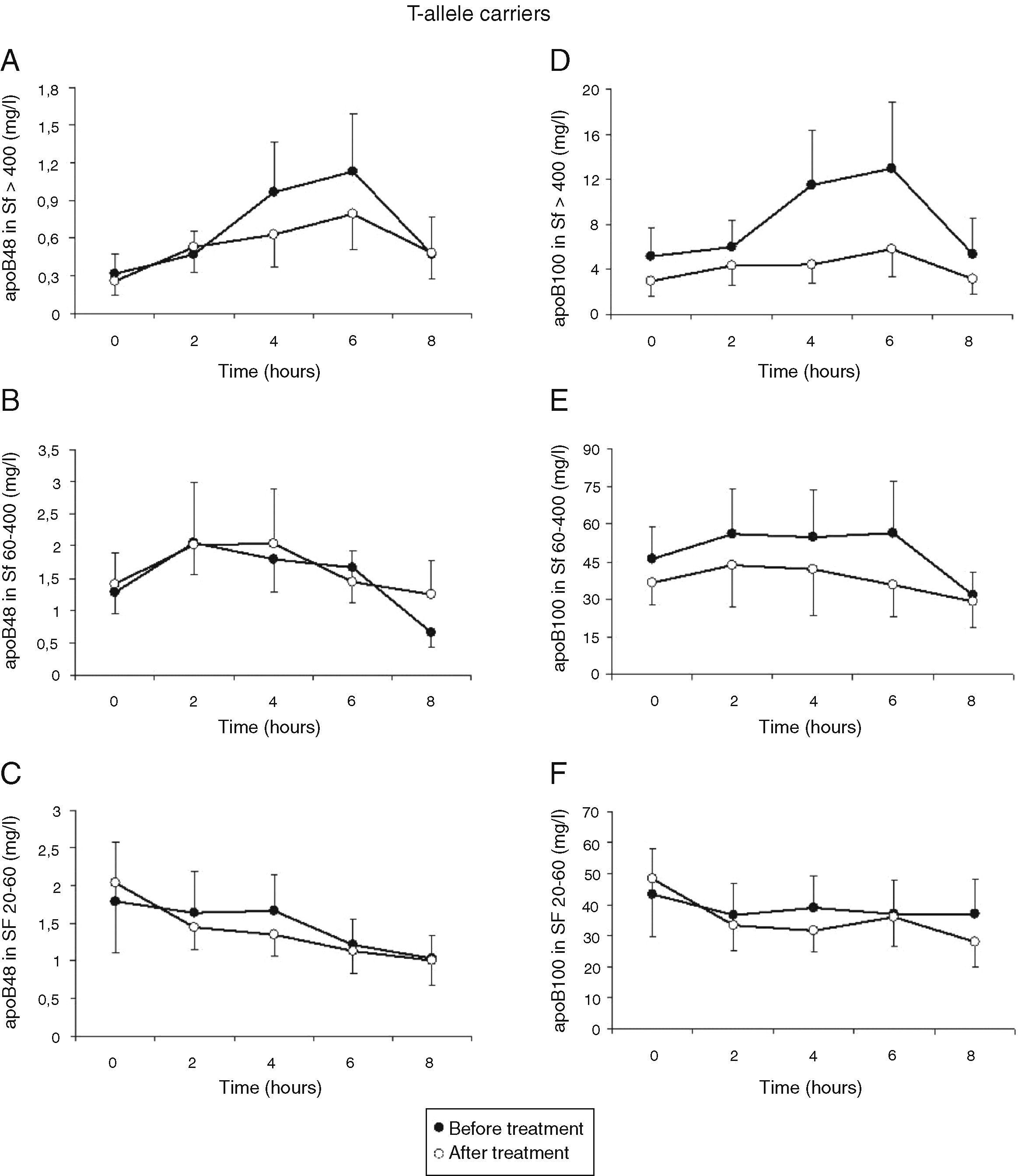
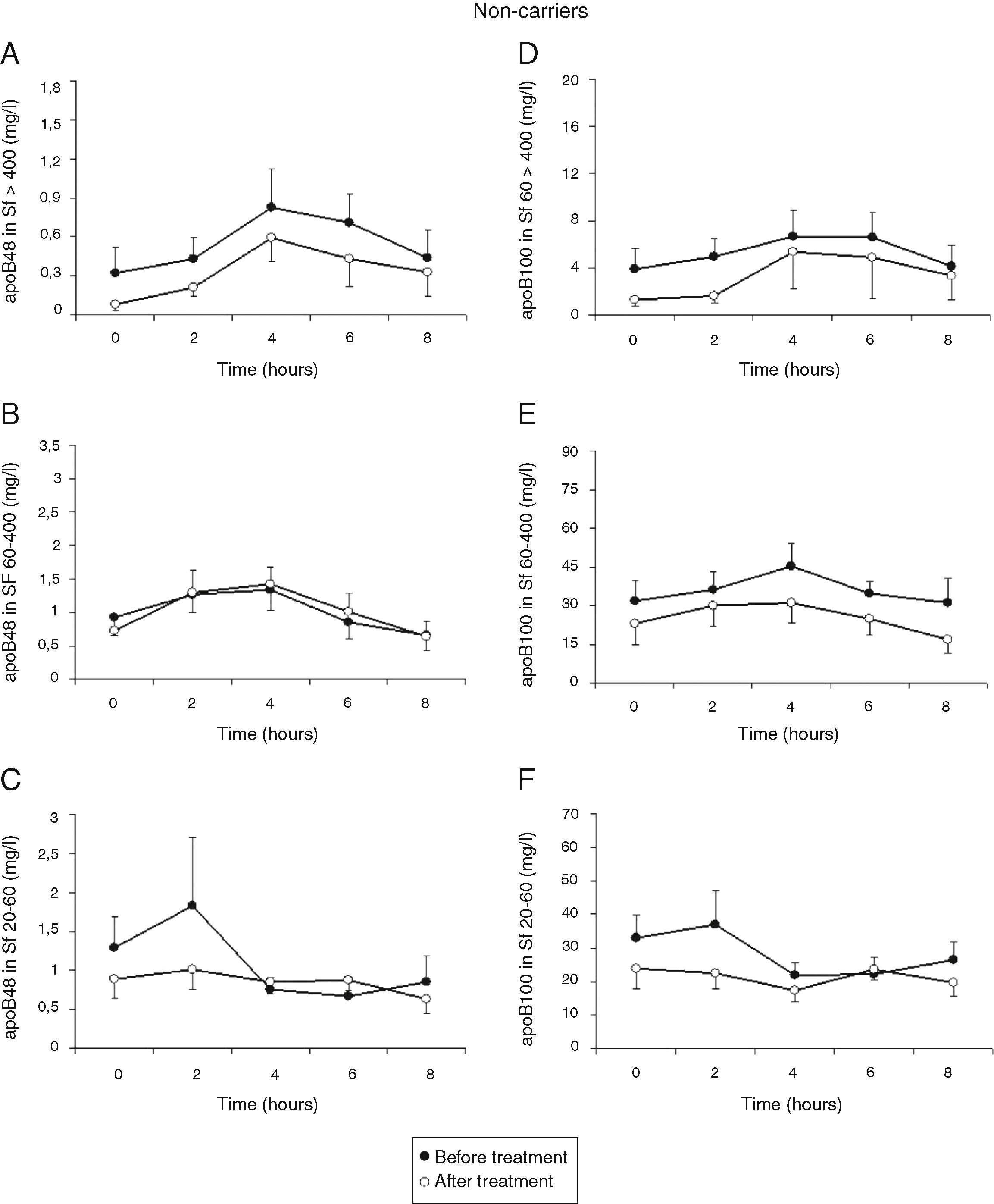
Treatment with atorvastatin resulted in a significantly lowered AUC for postprandial plasma TG in the T allele carriers (21.97±8.52mmolh/l vs 29.94±13.51mmolh/l, P=0.02), but postprandial triglyceridemia was unaltered in the non-carrier FCH patients (32.07±16.01mmolh/l vs 35.40±12.87mmolh/l, P=0.49), despite an approximately similar reduction in fasting TG (Fig. 3). The TG lowering effect of atorvastatin in non-carriers was most notable in the late postprandial phase (after 4–8h). Treatment with atorvastatin showed in both carriers of the T-allele and non-carriers a non-significant trend with lowered apo B48 in Sf >400 and lowered apo B100 in Sf >400 and Sf 60–400 (Figs. 4 and 5). Finally, the postprandial response for apo B100 in Sf 20–60 was significantly reduced after treatment with atorvastatin in non-carriers (221.7±106.4mgh/l vs 140.2±62.9mgh/l, P=0.04) (Fig. 5F).
 before (closed dots) and after treatment (open dots) with atorvastatin in FCH patients in non-carriers (A) and MTP−493T-allele carriers (B) during an oral fat load. The total area under the TG curve was significantly reduced after treatment in MTP−493T-allele carriers (P=0.02) but not in non-carriers.")
Mean postprandial changes of plasma triglycerides (TG) before (closed dots) and after treatment (open dots) with atorvastatin in FCH patients in non-carriers (A) and MTP−493T-allele carriers (B) during an oral fat load. The total area under the TG curve was significantly reduced after treatment in MTP−493T-allele carriers (P=0.02) but not in non-carriers.
 B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated (closed dots) FCH carriers of the MTP−493T-allele and after treatment with atorvastatin (open dots) during an oral fat load. No significant changes were found in the AUC of the different fractions for apo B48 and B100 after treatment. However, treatment with atorvastatin showed a trend with lowered apo B48 in Sf >400 and lowered apo B100 in Sf >400 and Sf 60–400.")
Mean postprandial changes of apolipoprotein (apo) B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated (closed dots) FCH carriers of the MTP−493T-allele and after treatment with atorvastatin (open dots) during an oral fat load. No significant changes were found in the AUC of the different fractions for apo B48 and B100 after treatment. However, treatment with atorvastatin showed a trend with lowered apo B48 in Sf >400 and lowered apo B100 in Sf >400 and Sf 60–400.
 B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated (closed dots) and after treatment with atorvastatin (open dots) in FCH non-carriers during an oral fat load. The AUC for apoB100 in Sf 20–60 was significantly reduced after treatment with atorvastatin (P=0.04). Additionally, treatment with atorvastatin showed a trend with lowered apo B48 in Sf >400 and lowered apo B100 in Sf >400 and Sf 60–400.")
Mean postprandial changes of apolipoprotein (apo) B48 and B100 in the Svedberg flotation fractions (Sf) >400 (A and D), 60–400 (B and E) and 20–60 (C and F) fraction in untreated (closed dots) and after treatment with atorvastatin (open dots) in FCH non-carriers during an oral fat load. The AUC for apoB100 in Sf 20–60 was significantly reduced after treatment with atorvastatin (P=0.04). Additionally, treatment with atorvastatin showed a trend with lowered apo B48 in Sf >400 and lowered apo B100 in Sf >400 and Sf 60–400.
Patients with FCH are characterized by exaggerated postprandial lipemia with a defective clearance of chylomicrons and their remnants. The major metabolic disturbance in FCH is a hepatic overproduction of hepatic TG-rich lipoproteins,30 but intestinal overproduction of chylomicrons has not been excluded yet.1 In other disorders with insulin resistance, for example type 2 diabetes mellitus, intestinal overproduction has also been proposed.31–33 The present pilot study suggests increased postprandial concentrations for apo B48 and B100 in the larger lipoproteins of untreated T-carriers. These results were not surprising since the T allele is known for its increased transcriptional activity of the MTP gene, which is associated with increased apo B48 concentrations.15,18,22 Our results indicate an increased number of smaller apo B containing lipoproteins in FCH patients with the T-allele, although the differences were not significant due to the small number of participants.
After treatment with atorvastatin, carriers of the T-allele showed a significant reduction in both fasting and postprandial TG in contrast to non-carriers. In addition, carriers of the T-allele showed a trend with a greater reduction in large VLDL (Sf >400) after treatment with atorvastatin. Statin therapy has been shown to lower intestinal MTP mRNA expression in diabetic and non-diabetic subjects.34 In vitro HepG2 cells showed a reduction in MTP mRNA transcription by approximately 50% when they were cultured in the presence of pravastatin to induce a sterol depleted condition.35 Since the T-allele is associated with higher transcriptional MTP activity,15 the statin inhibiting effect was probably more apparent in T-allele carriers.34
Our results show a more pronounced treatment response on postprandial lipemia in T-allele carrying FCH patients receiving atorvastatin. Two other studies have investigated the MTP−493G/T polymorphism in relation to treatment effects of lipid lowering therapy.21,22 Patients with heterozygous familial hypercholesterolemia showed a reduction in plasma TG after treatment with atorvastatin, but the TG reduction was dependent on the MTP−493G/T polymorphism.21 The T-allele was associated with a greater reduction in TG after treatment with atorvastatin, but only in males. In addition, a 3-month diet low on saturated fatty acids and high on monounsaturated and polyunsaturated fatty acids lowered TG, but its effect was more pronounced in carriers of the T-allele and especially in homozygous carriers of the T-allele.22 These results are in concordance with our findings.
MTP is required for the assembly of both intestinal and hepatic derived apo B containing lipoproteins. Despite similar postprandial TG levels between the different genotypes, increased plasma apo B48 concentrations have been found in the low Sf 20–60 fraction for homozygous carriers of the MTP−493T-allele in a previous study.18 We could not reproduce these differences in apo B48 concentrations between T-allele carrying and non-carrying FCH patients. However, we observed a trend of increased postprandial concentrations of apo B48 and B100 in the larger lipoproteins in carriers of the T-allele. This discrepancy between previous results and our study may be explained by the absence of homozygote carriers of the MTP−493T-allele in our study population or due to the limited number of study participants to reach conclusive results.
Several genes have been proposed to modulate the FCH phenotype,2,24 however MTP has not been found to contribute to the FCH phenotype in a previous study.24 This linkage study including 151 FCH patients out of 481 family members did not show an association between the MTP gene and the FCH phenotype in the fasting state, but they demonstrated a strong association between FCH and the apo AI-CIII-AIV gene cluster and the lecithin:cholesterol acyltransferase (LCAT) locus.24 Our data do not question the results of this study, but we did show subtle differences in FCH patients with a different MTP−493G/T polymorphism in relation to atorvastatin treatment.
In conclusion, a trend of increased postprandial apo B48 and B100 in FCH patients with the MTP−493T allele was observed compared to non-carriers. After treatment with atorvastatin FCH patients with the MTP−493T allele showed an increased reduction in fasting and postprandial TG compared to non-carriers of the variant allele.
Ethical responsibilitiesProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the responsible Clinical Research Ethics Committee and in accordance with those of the World Medical Association and the Helsinki Declaration.
Confidentiality of DataThe authors declare that they have followed the protocols of their work centre on the publication of patient data and that all the patients included in the study have received sufficient information and have given their informed consent in writing to participate in that study.
Right to privacy and informed consentThe authors have obtained the informed consent of the patients and /or subjects mentioned in the article. The author for correspondence is in possession of this document.
Conflict of interestThe authors declare that there are no conflicts of interest.
This study was supported by an unrestricted educational grant from Pfizer Inc and by the Research Foundation of the Department of Internal Medicine of the Sint Franciscus Gasthuis.