




To investigate the relationship between gut microbiota composition and the presence of coronary atherosclerosis assessed by coronary artery calcium (CAC) quantification in individuals without previous cardiovascular disease (CVD).
MethodsWe included 20 patients over 18 years of age with no history of CVD who underwent multiple detector-computed tomography (MDCT). From each patient, a stool sample was obtained to characterize gut microbiota composition by sequencing bacterial 16S ribosomal RNA gene. In addition, circulating levels of TNF-α and IL-1β, as well as trimethylamine N-oxide (TMAO) were determined in plasma samples by automated ELISA and capillary gas chromatography-mass spectrometry, respectively.
ResultsThe mean age of patients was 63.5 years and 60% were women. Half of patients had CAC > 100 (Agatston score), and were characterized by a higher abundance of the phylum Proteobacteria, mainly of bacteria belonging to the families Enterobacteriaceae, Pasteurellaceae, Erwiniaceae, Vibrionaceae and Morganellaceae, than patients with a CAC ≤ 100. Moreover, bacterial genera identified as biomarkers, such as Enterobacter, Escherichia/Shigella y Klebsiella, were positively associated with inflammation levels and with TMAO production.
ConclusionsOur data shows a gut microbiota profile associated with the presence of coronary calcium in patients without previous CVD. Although there are no strategies to decrease the amount of coronary calcium, gut microbiota is highly malleable by several factors. The possibility of preventing and even intervening CVD progression through strategies targeted gut microbiota is a very attractive idea that deserves further studies.
Determinar si existe alguna asociación entre el perfil de microbiota intestinal y la carga aterosclerótica global medida mediante cuantificación de calcio coronario (CCC) en sujetos sin antecedentes de enfermedad cardiovascular (ECV).
MétodosSe incluyeron 20 pacientes mayores de edad, sin antecedentes de enfermedad cardiovascular a los que se cuantificó el calcio coronario (CCC) mediante tomografía computarizada multicorte. Además, se les recogió una muestra de heces para caracterizar la composición de la microbiota intestinal mediante la secuenciación del gen 16S RNAr con técnicas de secuenciación masiva y una muestra de sangre para la cuantificación de citoquinas proinflamatorias, mediante ELISAs específicos, y del metabolito bacteriano N-óxido de trimetilamina (TMAO) por cromatografía de gases/líquidos acoplada a espectrometría de masas en tándem.
ResultadosLa media de edad fue de 63,5 años y un 60% eran mujeres. La mitad de los pacientes (n = 10) presentaron una CCC > 100 y se caracterizaron por una mayor abundancia de bacterias del filo Proteobacteria, pertenecientes principalmente a las familias Enterobacteriaceae y Pasteurellaceae, que los pacientes con CCC ≤ 100. La mayoría de los géneros bacterianos identificados Enterobacter, Escherichia/Shigella, Klebsiella, Citrobacter y Salmonella se asociaron positivamente con los niveles plasmáticos de TNF-α o IL-1β y con la producción de TMAO.
ConclusiónLos resultados de este estudio piloto muestran un perfil de microbiota intestinal asociado a la presencia de CCC > 100 en pacientes sin ECV previa, caracterizado por un aumento en la proporción de géneros bacterianos productores de TMAO. Puesto que la composición de la microbiota intestinal es altamente modulable por diversos factores, es posible que, en un futuro, podamos prevenir, e incluso intervenir, la ECV mediante estrategias nutricionales.
Screening for subclinical atherosclerotic disease is a medical challenge, as more than half of initial coronary events, including sudden death, occur in patients who have not previously experienced any symptoms.1 In recent years great interest has therefore arisen in developing new strategies to identify individuals who are at higher cardiovascular risk. The image-based quantification of coronary calcium using computed axial tomography (CAT) is now considered to be most precise tool for the selection of patients with coronary atherosclerosis,2,3 as it is a non-invasive technique that gives rapid results, and patients do not have to interrupt their medication for it to be used.
The basic lesion in arteriosclerosis is plaque, which is characterized by being an accumulation of lipids, fibrous tissue, calcium and inflammatory cells on the vascular wall.4 Coronary calcification starts in the first phases of life, but it progresses faster in older subjects who have more advanced atherosclerotic lesions. It only occurs in arteries that have already been damaged, and does not appear in normal arteries.5 Although an absence of coronary calcium does not exclude the existence of non-calcified plaque,6,7 the presence of calcified coronary arteries increases the probability of ischemic disease by more than ten times in asymptomatic individuals; a probability that is substantially higher than those associated with conventional risk factors.7 In fact, North American and European clinical guides consider the calcium score to be a very useful tool when evaluating and stratifying cardiovascular risk in asymptomatic individuals, and it may be used as a guide to manage future preventive therapies.8,9
Our organism is inhabited by millions of microorganisms, chiefly bacteria, which form the human microbiota.10 They live mainly in the gut and, in some way during the evolutionary process, a symbiotic relationship has come into being between humans and their microbiota. In recent years many chronic diseases, including cardiovascular diseases (CVD), have been linked to changes in the composition of the gut microbiota (dysbiosis). Thus several studies have shown that an association exists, not only between gut dysbiosis and atherosclerosis, hypertension, heart failure, chronic renal disease or obesity/type 2 diabetes mellitus, but also with their clinical complications.11 A study by Liu et al. proved that changes in the composition of gut microbiota were able to precisely distinguish patients with stable coronary arterial disease from those with acute coronary syndrome.12 An experimental model of ischemia/reperfusion demonstrated that an alteration in the gut microbiota induced by an antibiotic or a probiotic led to smaller infarcts and better recovery of post-ischemic mechanical function in comparison with untreated animals.13 Recent results in our group of an experimental model of heart failure (HF) showed that changes in the composition of the gut microbiota occurred prior to the manifestation of cardiac alterations, and that they were associated with the evolution of structural and functional changes in the heart.14 What is more, the gut microbiota functions as a host endocrine system, and it may contribute to the development of CVD by freeing metabolites which influence inflammation, dyslipidaemia, vascular tone control or fibrosis.11 Taken together, these results suggest that intestinal dysbiosis may be a major risk factor for the development of arteriosclerosis.
The aim of this work was therefore to perform a pilot study, to investigate the relationship between the composition of the gut microbiota and cardiovascular risk in individuals without previous cardiovascular disease, according to the calcification of their coronary arteries.
Material and methodsStudy populationAdult patients were included who had no history of cardiovascular disease (HF, coronary arterial disease or ictus). They were subjected to a multidetector CAT to quantify their coronary calcium in Hospital Universitario Río Hortega, Valladolid, due to the existence of symptoms of thoracic pain with no clear ischemic profile, without definitive functional tests and with a low pre-test probability of coronary disease. Patients with deteriorated renal or hepatic function were excluded, as were those with cancer, inflammatory diseases, previously diagnosed clinical or subclinical atherothrombótic arterial disease or treatment with antibiotics or probiotics in the two months prior to their inclusion. Anthropometric and biochemical data were recorded for all of the participants, together with their cardiovascular risk factors. Cardiovascular risk factors were evaluated using scores on the Framingham scale calibrated for the Spanish population (REGICOR). This evaluated the risk of suffering coronary disease (myocardial infarct or death) in the following 10 years. Patients were classified as low coronary risk (0%–9%), moderate (10%–19%) or high (≥20%).15
This work was undertaken according to the fundamental principles set in the Helsinki Declaration, the Council of Europe Agreement on human rights and biomedicine, and the Universal Declaration of UNESCO on the human genome and human rights. All of the subjects gave their informed consent, and the project was approved by the Ethics Committee of Hospital Clínico San Carlos (C.I. 16/237-E).
Coronary calcium quantificationCoronary calcium was quantified for all of the patients by multidetector CAT (128 detectors, Siemens Somaton Drive, Siemens Healthcare GmbH, Erlangen, Germany). The study protocol included prospective acquisition based on an electrocardiogram (ECG) (ECG-gating) in one beat and without prior administration of contrast of the whole cardiac volume, with subsequent reconstruction of the images obtained (3.0 mm slice thickness). The post-process analysis of the images was performed using specific software (SYNGO VIA, Siemens Healthcare GmbH), and they were interpreted by two independent observers. The Agatston calcium score was calculated for each lesion, based on the area and density of the calcified lesions according to the method described,16 and the total was calculated to determine its value for each patient.
The collection of faecal samples and extraction of microbial DNAA sample of faeces was obtained from each individual in a tube with DNA stabilizer (OMNIgene-GUT, DNAgenotek, Ottawa, Canada) and it was stored at −80 °C until it was processed. The microbial DNA of the samples was extracted using the QIAamp Fast DNA Stool (Quiagen, Hilden, Germany) mini-kit following the manufacturer’s instructions. DNA integrity was evaluated using a BioAnalyzer 2100 (Agilent, Palo Alto, CA, U.S.A.), and its concentration was determined using a Qubit 3.0 fluorimeter with the dsDNA HS assay (Thermo Fisher Scientific, Carlsbad, Ca, U.S.A.).
16S ribosomal RNA gene sequencingFor each faecal sample the 16S ribosomal RNA gene was amplified using the Ion 16S Metagenomics (Thermo Fisher Scientific) kit to amplify 7 hypervariable regions (V2, V3, V4, V6-7, V8 and V9) of the said gene. The libraries were prepared using 5 ng DNA per sample, converting the ends of the amplicons into blunt-ends using the Ion Plus Fragment Library (Thermo Fisher Scientific) kit and adding molecular DNA identifiers using the Ion Express Barcode Adapters kit (Thermo Fisher Scientific). The libraries were then diluted to 22 pM, they were subjected to PCR clonal amplification in emulsion using the Ion OneTouch™ 2 System (Thermo Fisher Scientific), and they were sequenced in Ion S5™ System apparatus using an Ion 520™ Chip (Thermo Fisher Scientific).
Bioinformatic analysisThe sequences obtained were automatically pre-processed by Torrent Suite™ software (v5.10.0; Thermo Fisher Scientific). The regions of the primers were eliminated and the sequences were cut to 150 pairs of bases using Python. The FASTQ files were analysed using QIIME 2 (v2020.2) software to dereplicate the readings and eliminate the sequences that only appeared once (singletons). The final sequences were grouped in taxonomic units (OTU) with 99% similarity using the SILVA (v138) database. The taxonomic assignation of the identified OTU was performed using QIIME 2 and SILVA taxonomy. Bacterial species richness (alpha diversity) was measured using a rarefied OTU profile at a sequencing depth of 115,435 readings. The relative abundance of the taxons was calculated as a percentage based on the total number of readings per sample. The Bray-Curtis index was calculated to study beta diversity. Principle coordinate analysis (PCoA) was applied to the resulting dissimilarity matrix and bidimensional PCoA graphs were used to visualize the compositional structure between groups and samples. The PICURSt2 (v2.2.0-b) software package was used to predict the functional content of the microbial communities as the orthologues profile of the Kyoto Encyclopaedia of Genes and Genomes (KEGG). The size of the effect of lineal discriminatory analysis (LEfSe) was used to identify the taxons and differentially abundant functional routes between the groups, using the scripts shared by the author (github.com/SegataLab/lefse). Taxons with a relative abundance <0,01% were eliminated for this analysis and the results were selected with a LDA score ≥2.0 and P < .05. The analytical protocol had been published in greater detail beforehand by our group.14
Obtaining plasma and quantifying proinflammatory cytokinesA sample of peripheral blood was obtained from each patient by venepuncture in vacuum tubes with EDTA as the anticoagulant. Plasma was obtained by centrifuging the blood at 1500 rpm during 10 min. The samples were then divided into aliquots and stored at −80 °C.
Circulating levels of TNF-α and IL-1β were determined in plasma by automatic ELISA (Protein Simple Plex Assay, Biotechne, MN, U.S.A.) following the manufacturer’s instructions. The sensitivity of the TNF-α assay was 0.278 pg/mL. The intra- and inter-assay variation coefficients were 5.0% and 0.771%, respectively. The sensitivity of the IL-1β assay was 0.064 pg/mL. The intra- and inter-assay variation coefficients were 4.9% and 0.342%, respectively.
Quantification of the TMAO bacterial metaboliteTrimethylamine N-oxide (TMAO) detection was carried out in plasma samples by liquid chromatography coupled to a mass spectrometer (LC-MS) using an electrospray ionization system (ESI) with a simple quadrupole (Qp) as analyser. Plasma samples were deproteinized using methanol as the precipitating reagent and they were centrifuged at 12,500 rpm during 2 min. at ambient temperature. Chromatographic separation was then performed by injecting 10 μL of supernatant in an LC-MS 2020 system (Shimazdu Europa GmbH, Duisburg, Germany) equipped with a C18 (150 mm × 4.6 mm × 5 μm) Supelco reverse phase column (Merck Life Science SLU, Madrid, Spain) at 35 °C. Elution was performed in a gradient with an aqueous solution of formic acid 1N/methanol during 14 min at a rate of flow of 0.2 mL/min. TMAO was detected using a simple quadrupole with nitrogen as the nebulization gas (1.5 L/min flow) and drying gas (10 L/min flow) produced by a generator (Peak Scientific Spain SL, Madrid, Spain). To carry out quantification the ratio of heights between the TMAO and the internal standard was used, monitoring the molecular ions (TMAO: m/Z = 76 y D9-TMAO: m/Z = 85) in Selected Ion Monitoring mode. To evaluate the parameters of linearity, exactitude and recovery 6 duplicate concentrations of TMAO were prepared (Sigma-Aldrich Química S.A., Madrid, Spain) of 1, 5, 10, 25, 50 and 100 μmoles/l. The linearity of the method was analysed by lineal regression.
Statistical analysisThe variables included in the study, as well as alpha diversity and the abundance of the different taxons, were expressed as averages ± standard deviation from the average or median (interquartile range). The Shapiro-Wilk test was used to check the normality of each variable. Those with a normal distribution were compared statistically using the Student’s t-test, and those which followed a non-normal distribution were compared using the non-parametric Mann-Whitney U test. Qualitative variables were analysed using Fisher’s exact test. Differences between groups respecting the structure of bacterial communities (beta diversity) were analysed using permutation analysis and multiple ANOVA (PERMANOVA) with 999 permutations. The association between the relative abundancies of genera and plasma levels of TNF-α and IL-β was analysed using Spearman correlations. Statistical analysis was undertaken using the Python SciPy package. For all of the analyses differences were considered to be statistically significant if P < .05.
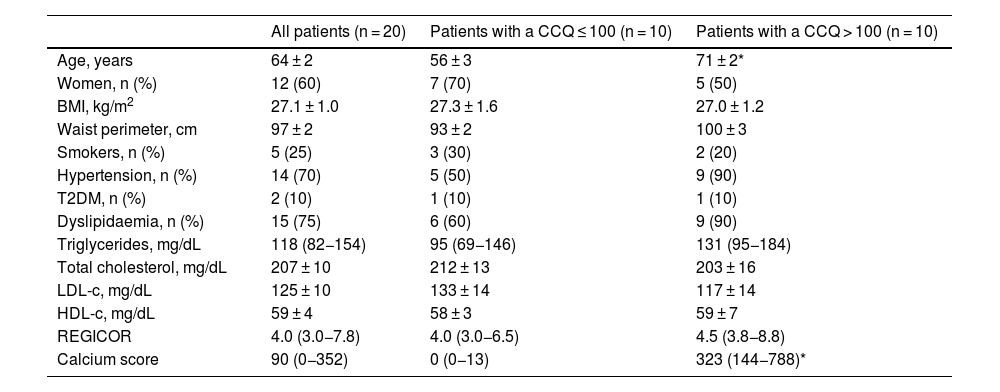
ResultsClinical characteristics of the patientsA total of 20 subjects were included in this pilot study. They had all been subjected to a multidetector CAT prescribed by their doctor to rule out the presence of ischemic cardiac pathology. Their average age was 63.5 years, and 60% were women. According to the REGICOR equation, 15 patients had a low risk of suffering a cardiovascular event, and two had a moderate risk, with an average risk of 5.0%. Cardiovascular risk could not be calculated for three patients because they were over 74 years old. Nevertheless, 10 patients had a CCQ > 100, with a high probability of coronary disease and high cardiovascular risk, as opposed to 10 patients who had a CCQ ≤ 100, with a minimum to slight probability of coronary stenosis and low to moderate cardiovascular risk. The patients with a CCQ > 100 were significantly older, with a slight tendency towards a greater presence of hypertension and dyslipidaemia than was the case in the patients with a CCQ ≤ 100, although there were no significant differences between both groups in cardiovascular risk according to the REGICOR equation (4.5 [3.8−8.8] vs. 4.0 [3.0−6.5], P = NS). The anthropometric, clinical and biochemical characteristics of the included subjects shown in Table 1 according to their CCQ.
Clinical and biochemical characteristics of all the patients according to their coronary calcium quantification (CCQ).
| All patients (n = 20) | Patients with a CCQ ≤ 100 (n = 10) | Patients with a CCQ > 100 (n = 10) | |
|---|---|---|---|
| Age, years | 64 ± 2 | 56 ± 3 | 71 ± 2* |
| Women, n (%) | 12 (60) | 7 (70) | 5 (50) |
| BMI, kg/m2 | 27.1 ± 1.0 | 27.3 ± 1.6 | 27.0 ± 1.2 |
| Waist perimeter, cm | 97 ± 2 | 93 ± 2 | 100 ± 3 |
| Smokers, n (%) | 5 (25) | 3 (30) | 2 (20) |
| Hypertension, n (%) | 14 (70) | 5 (50) | 9 (90) |
| T2DM, n (%) | 2 (10) | 1 (10) | 1 (10) |
| Dyslipidaemia, n (%) | 15 (75) | 6 (60) | 9 (90) |
| Triglycerides, mg/dL | 118 (82−154) | 95 (69−146) | 131 (95−184) |
| Total cholesterol, mg/dL | 207 ± 10 | 212 ± 13 | 203 ± 16 |
| LDL-c, mg/dL | 125 ± 10 | 133 ± 14 | 117 ± 14 |
| HDL-c, mg/dL | 59 ± 4 | 58 ± 3 | 59 ± 7 |
| REGICOR | 4.0 (3.0−7.8) | 4.0 (3.0−6.5) | 4.5 (3.8−8.8) |
| Calcium score | 90 (0−352) | 0 (0−13) | 323 (144−788)* |
Values are expressed as an average ± SD or median (interquartile range) for continuous variables or n (%) for categorical variables.
HDL-c: HDL cholesterol; LDL-c: LDL cholesterol; T2DM: type 2 diabetes mellitus; BMI: body mass index.
Sample sequencing identified a total of 4,457,899 readings that were classified in 21,092 OTUs. The average number of readings in the patients with a CCQ ≤ 100 was 225,515, and 220,275 in the patients with a CCQ > 100.
To analyse the composition of the gut microbiota, we first calculated the alpha diversity of the samples. This parameter takes into account the intrinsic biodiversity of each sample. We found no statistically significant differences in terms of bacterial richness (expressed by rarefication curves) (Appendix B, supplementary figure 1A of the annex available online) or in the Shannon index (a parameter that takes the richness and the distribution of the OTUs into account) (Appendix B, supplementary figure 1B) in samples from patients with a CCQ ≤ 100 in comparison with patients with a CCQ > 100. Analysis of beta diversity in the gut microbiota samples showed that the samples tended to be grouped in one of two groups, depending on their calcium quantification. However, this did not attain statistical significance (Appendix B, supplementary figure 1C).
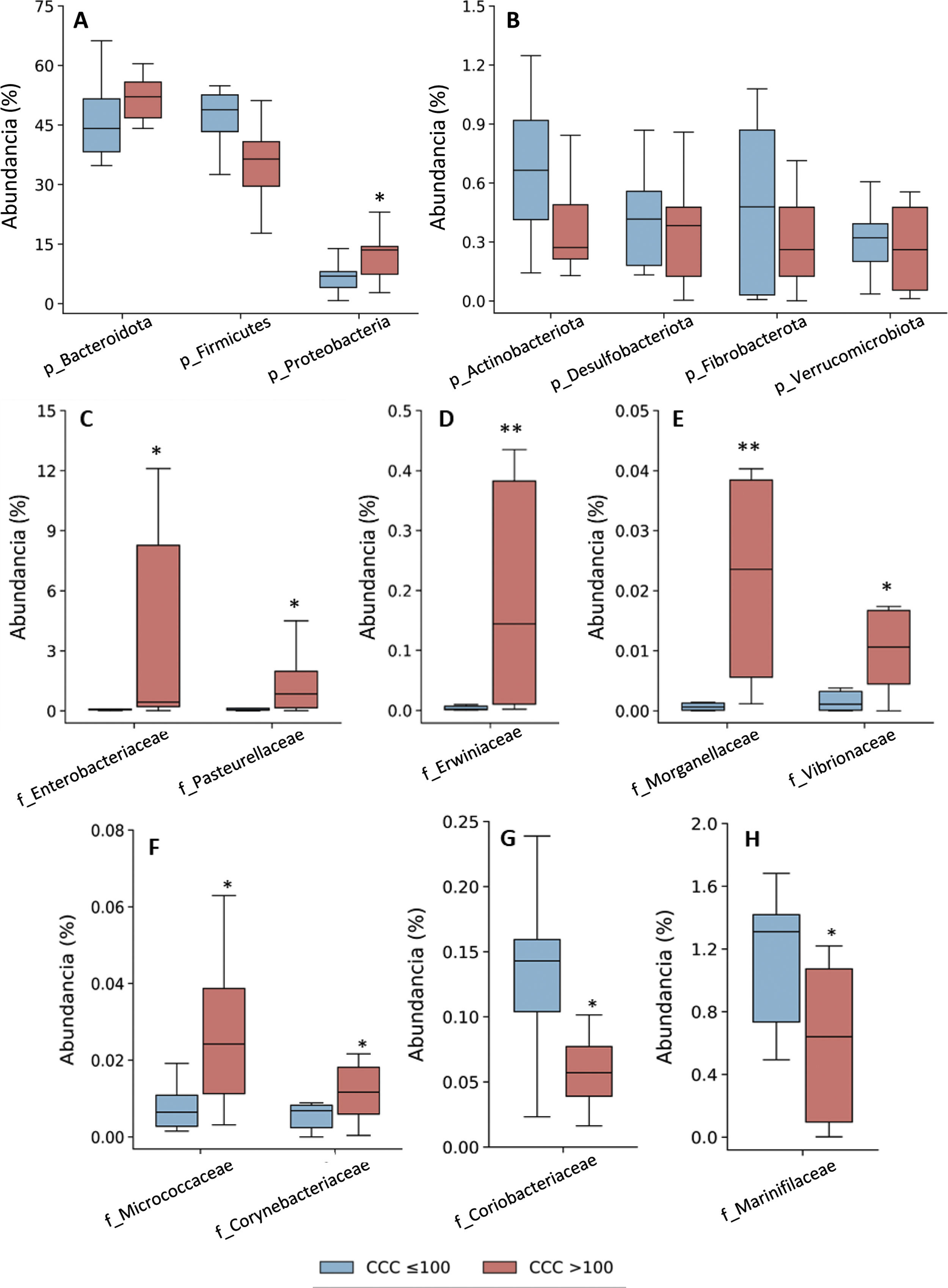
Taxonomic assignation of the OTU led to the identification of 18 phyla, 27 classes, 52 orders, 115 families and 408 genera, taking into account all of the patients included in the study. The bacterial composition in patients with a CCQ ≤ 100 was similar to those of the patients with a CCQ > 100, although differences were observed in the distribution of the taxons. In both groups the most abundant phyla were Firmicutes, Bacteroidota (previously Bacteroidetes) and Proteobacteria, corresponding to more than 97% of the bacteria that were identified, although they differed in abundance between the groups; in the patients with a CCQ > 100, there were more bacteria in the phyla Bacteroidota and Proteobacteria, while there were less of Firmicutes than in the patients without coronary calcium (Fig. 1). On the other hand, in both groups approximately 3% of the bacteria belonged to minority phyla, although no significant differences were detected between the groups in this respect (Fig. 1).
 and relative abundance of bacteria belonging to the families of the Proteobacteria (C–H) phylum. The results are shown in box diagrams with the median, interquartile range and maximum and minimum values. f: family; p: phylum. *P < .05 vs. patients with a CCQ ≤ 100. **P < .01 vs. patients with a CCQ ≤ 100.")
Taxonomic composition of the gut microbiota at phylum level (A and B) and relative abundance of bacteria belonging to the families of the Proteobacteria (C–H) phylum. The results are shown in box diagrams with the median, interquartile range and maximum and minimum values.
f: family; p: phylum.
*P < .05 vs. patients with a CCQ ≤ 100.
**P < .01 vs. patients with a CCQ ≤ 100.
Given that there was a significant age difference between both groups of patients, we analysed the abundance of proteobacteria according to patient age (older or younger than 74 years). In the group of patients younger than 74 years, those with a CCQ > 100 showed a greater abundance of proteobacteria than the patients with a CCQ ≤ 100 (Appendix B, supplementary figure 2). In the group of patients older than 73 years, all of the patients had a CCQ > 100, although no significant differences were found in the amount of proteobacteria respecting the patients under 74 years (Appendix B, supplementary figure 2).
As a result of the increase in proteobacteria, a greater abundance of bacteria of the class Gammaproteobacteria was observed (phylum Proteobacteria) (CCQ ≤ 100: 3.8% [2.8–7.0]; CCQ > 100: 13.5% [7.4–14.4], P < .05). At family level the bacteria in the families Enterobacteriaceae, Pasteurellaceae, Erwiniaceae, Vibrionaceae and Morganellaceae (Gammaproteobacteria class), Micrococcaceae and Corynebacteriaceae (Actinobacteriota -previously Actinobacteria class) were significantly more abundant in patients with a CCQ > 100 than they were in those with a CCQ ≤ 100 (Fig. 1C–F). On the contrary, the families Coriobacteriaceae (Actinobacteriota class) and Mariniphylaceae (Bacteroidota class) were significantly less abundant in patients with CCQ > 100 (Fig. 1G,H).
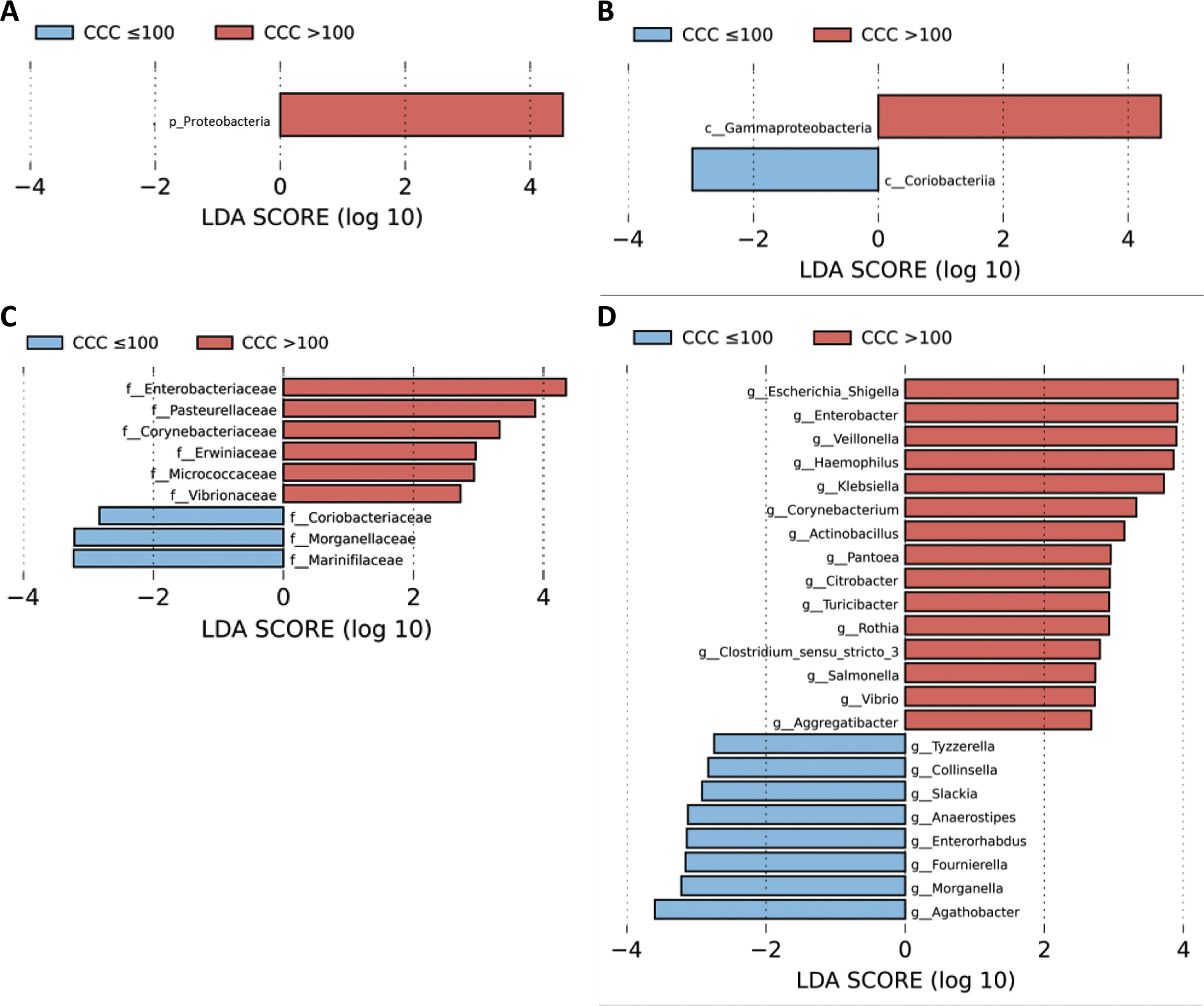
A LEfSe analysis was then performed on all of the taxonomic levels. This bioinformatics tool allows us to infer whether any taxon may function as a biomarker of patients with a high level of cardiovascular risk according to their CCQ. The Proteobacteria phylum, its class Gammaproteobacteria and the families of this class, Enterobacteriaceae, Pasteurellaceae, Erwiniaceae and Vibrionaceae were identified as taxonomic biomarkers (LDA score ≥ 2.0 and P < .05) between patients with and without coronary calcium (Fig. 2A–C). At genus level the identification of taxonomic biomarkers showed that fifteen genera were significantly more abundant in patients with a CCQ > 100 (LDA score ≥ 2.0 and P < .05), of which ten belonged to the phyla Proteobacteria: Escherichia/Shigella, Enterobacter, Klebsiella, Citrobacter, Salmonella (Enterobacteriaceae family), Haemophilus, Actinobacillus, Aggregatibacter (Pasteurellaceae family), Pantoea (Erwiniaceae family) and Vibrio (Vibrionaceae family) (Fig. 2D).
, classes (B), families (C) and genera (D) identified as biomarkers in patients with a CCQ > 100 in comparison with patients with a CCQ ≤ 100. The length of the horizontal bars represents the LDA score. P < .05; LDA score ≥ 2.0. c: class; f: family; g: genus; p: phylum.")
LEfSe analysis showing phyla (A), classes (B), families (C) and genera (D) identified as biomarkers in patients with a CCQ > 100 in comparison with patients with a CCQ ≤ 100. The length of the horizontal bars represents the LDA score. P < .05; LDA score ≥ 2.0. c: class; f: family; g: genus; p: phylum.
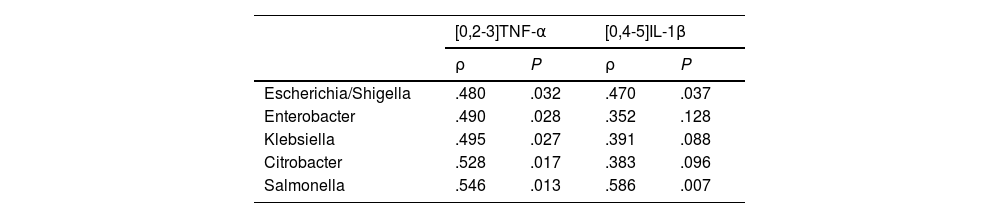
The patients with a CCQ >100 had higher levels in plasma of TNF-α than the patients with a CCQ ≤ 100 (6.3 ± 0.53 vs. 4.5 ± 0.19 pg/mL, P < .05). All of the genera in the Enterobacteriaceae family that were identified as biomarkers of the patients with a CCQ > 100 correlated positively with TNF-α levels (Table 2). Although no significant differences were found between levels of IL-1β in plasma between both groups of patients (.074 ± .034 vs. .066 ± .023 pg/mL, P = NS), the abundance of two bacterial genera (Escherichia/Shigella and Salmonella) was also associated with a higher concentration of IL-1β (Table 2).
Correlation between circulating levels of TNF-α and IL-1β, and Enterobacteriaceae family bacteria.
| [0,2-3]TNF-α | [0,4-5]IL-1β | |||
|---|---|---|---|---|
| ρ | P | ρ | P | |
| Escherichia/Shigella | .480 | .032 | .470 | .037 |
| Enterobacter | .490 | .028 | .352 | .128 |
| Klebsiella | .495 | .027 | .391 | .088 |
| Citrobacter | .528 | .017 | .383 | .096 |
| Salmonella | .546 | .013 | .586 | .007 |
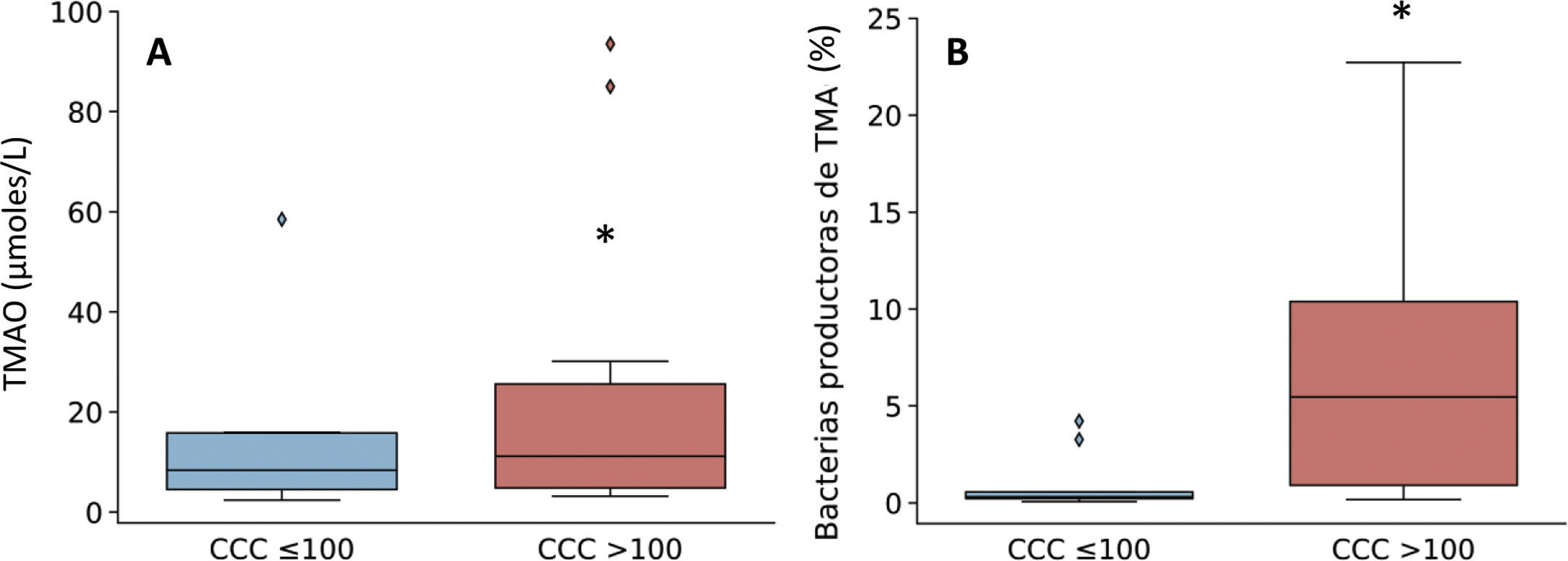
Given that the patients with a CCQ > 100 were characterized by an increase in the genera of bacteria that produce TMA, such as Enterobacter, Escherichia/Shigella and Klebsiella, the production of TMAO was then quantified. This is a bacterial metabolite which originates in TMA, and it is considered to be a cardiovascular risk factor.11 As can be seen in Fig. 3, levels in plasma of TMAO were raised in subjects with a CCQ > 100 and positively correlated with the amount of calcium (R = .54; P = .017).
 and relative abundance of TMA-producing bacterial genera (B) according to the presence of coronary calcium. The results are shown in box diagrams with the median, interquartile range and maximum and minimum values. *P < .05 vs. patients with a CCQ ≤ 100.")
Levels in plasma of TMAO (A) and relative abundance of TMA-producing bacterial genera (B) according to the presence of coronary calcium. The results are shown in box diagrams with the median, interquartile range and maximum and minimum values. *P < .05 vs. patients with a CCQ ≤ 100.
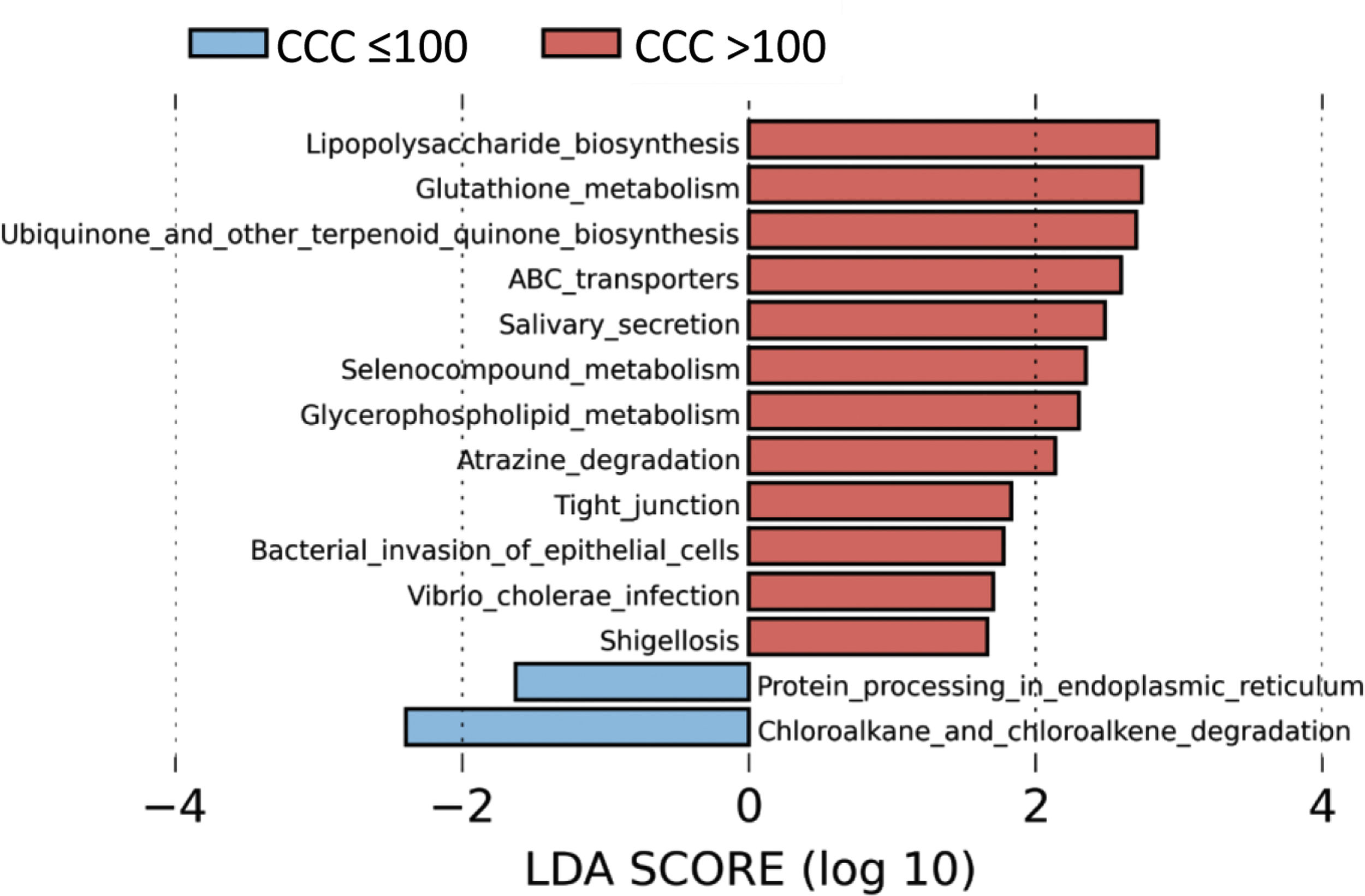
To identify the functional profile of the gut microbiota in patients with and without coronary calcium, we undertook a LEfSe analysis to compare the KEGG metabolic routes of the patients with a CCQ > 100 in comparison with those who had a CCQ ≤ 100. We identified 12 increased routes in the patients with a CCQ > 100 in comparison with the calcium-free patients. These routes were mainly associated with lipopolysaccharide synthesis (LPS), antioxidant mechanisms such as glutathione metabolism or ubiquinone synthesis, or ones associated with bacterial infections or invasion of the epithelium by microorganisms (Fig. 4).
Discussion
This work identified a gut microbiota profile that is associated with the presence of coronary calcium in patients with no previous cardiovascular disease. Patients with a calcium score >100 are characterized by having a greater abundance of proteobacteria, chiefly those in the Enterobacteriaceae and Pasteurellaceae families, in comparison with patients with a calcium score ≤100. The majority of the bacteria genera identified are positively associated with the levels of inflammation in patients and with production of the bacterial metabolite TMAO.
The presence of coronary calcium has been considered for years to be a direct marker of the existence of atherosclerosis, and numerous studies have demonstrated its usefulness in stratifying the degree of cardiovascular risk, even in individuals classified as low risk but with risk functions.2,3 In this work half of the patients without previous cardiovascular disease had coronary calcium, a result which is very similar to the one described by other authors in a Spanish population.17 Nevertheless, although coronary CAT is a non-invasive technique, it is only available to a limited extent and involves a certain exposure to radiation. There is therefore a high level of interest in identifying more convenient biomarkers. Metagenomic analysis of faecal samples from patients allowed us to identify a gut microbiota profile that is characterized by a predominance of bacteria in the Proteobacteria phylum and more specifically the Enterobacteriaceae family in individuals with coronary calcium. As far as we know this is the first work to analyse the composition of gut microbiota in association with the existence of coronary calcium in patients without known CVD. However, our results agree with those of previous studies which investigated gut microbiota composition in patients with symptomatic atherosclerosis. In different populations of patients with atherosclerotic CVD (stable or instable angina or acute myocardial infarct) a significant increase in the Enterobacteriaceae family has been reported, mainly of the Escherichia-Shigella, Klebsiella and Enterobacter genera.11 It has also been reported that the increase in proteobacteria such as Klebsiella, Streptococcus, Haemophilus and Granulicatella, as well as the production of bacterial metabolites, correlates positively with the severity of CVD.12 Another genus of bacteria that was identified, Citrobacter, has been associated with greater carotid íntima-media thickness in a population highly susceptible to developing cardiovascular disease.18 Furthermore, although the quantities were not large, some studies have identified the presence of enterobacteria in atheromatous plaques, chiefly Klebsiella, Pantoea, Citrobacter or Enterobacter, which are also found in the oral microbiota and intestines.19,20
Inflammation is well-known to play a fundamental role in the commencement, development and complications of arteriosclerosis.4 There is in fact a close relationship between inflammatory markers such as TNF-α, IL-1β, IL-6 or C-reactive protein (CRP) and the risk of suffering future cardiovascular events, even in apparently healthy subjects.4 Previous studies suggest that the information supplied by inflammatory markers in predicting the risk of coronary heart disease would be complementary to the information provided by CCQ.21 In recent years the origin of this inflammatory state has been associated with the gut microbiota. LPS is one of the main components of the wall of gram-negative bacteria, and this may trigger an intense inflammatory response when it reaches the circulation.11 There is strong evidence that the connection between LPS-mediated low grade inflammation and the development of metabolic and inflammatory disorders.22,23 The Enterobacteriaceae family are gram negative bacteria. The inoculation of germ-free mice with bacteria of the Enterobacter genus (Enterobacteriaceae family) taken from obese patients has been shown to induce a significant weight increase in the animals, as well as several inflammatory markers.24 The increase in enterobacteria is often observed in intestinal dysbiosis associated with numerous pathologies which involve intestinal inflammation, and different mechanisms have been suggested for this.25,26 On one hand, while under normal conditions the epithelial cells in the colon consume the oxygen of the lumen and create an anaerobic atmosphere, when there is intestinal inflammation the colonocytes modify their metabolism to anaerobic glycolysis. As this consumes less oxygen it increases its availability and favours the growth of optional anaerobic micro-organisms such as enterobacteria.25 Another important factor that may be connected with this is the respiratory flexibility of enterobacteria, as they are able to use the nitrate generated in inflammatory states as an alternative electron acceptor.25 The lack of butyrate has been described to induce a change in colonocyte metabolism to anaerobic glycolysis on the one hand, and greater expression of the coding gene for nitric oxide synthase (NOS) on the other, giving rise to higher nitrate production.27 This suggests a mechanism in which the absence of a healthy gut microbiota which produces butyrate may lead to excessive growth of bacteria in the Enterobacteriaceae family.
In our work we found an association between the abundance of Enterobacteriaceae family genera such as Enterobacter, Escherichia/Shigella and Klebsiella, and levels in plasma of TMAO in patients with a CCQ > 100. The Enterobacteriaceae family are the main producers of trimethylamine (TMA),28 the precursor metabolite of TMAO, associated with the development of atherosclerosis and coronary heart disease.11 Several studies have shown that TMAO promotes the adherence of monocytes to the endothelium and activates protein kinase-C routes and the nuclear κB (NF-κB) factor, which induces endothelial dysfunction and accelerate the atherosclerotic process.11,29 Moreover, high levels of TMAO increase platelet response, promoting thrombosis.29,30 A study with more than 4000 participants showed that the concentration of TMAO in plasma may predict the risk at 3 years of a thrombotic event (cardiac infarction or cerebrovascular accident).30
The predicted metabolic routes associated with the gut microbiota identified a significant increase in LPS biosynthesis routes in patients with a CCQ > 100, consistent with the abundance of bacteria in the Enterobacteriaceae family. Furthermore, patients with a CCQ > 100 showed an increase in the genes involved in glutathione metabolism, which protects against oxidative stress, especially in the heart. Thus alterations in the homeostasis of the cardiac glutathione metabolism have been associated with anomalies in cardiac function and structure.31 We also see a significant rise in the route associated with the biosynthesis of ubiquinone, which has been associated with a lower degree of inflammation and oxidative stress in patients with ischemic myocardiopathy.32 It is possible that the increase in glutathione and ubiquinone metabolism in patients with a CCQ > 100 is a compensatory mechanism. We also see an increase in the routes associated with infection by bacteria in the Enterobacteriaceae family in epithelial cells, such as Shigellosis or infection by Vibrio cholerae.
The patients with a larger amount of coronary calcium were significantly older than the ones with less calcium in their arteries. The gut microbiota is a dynamic ecosystem which is influenced by numerous factors, and its composition is well-known to change depending on age.33 At phylum level, Firmicutes predominates in adults and falls in the elderly, while there is a certain degree of disagreement about whether the Bacteroidota phylum decreases or increases with age. On the contrary, there is agreement about the rise in levels of bacteria in the Proteobacteria phylum, especially the Enterobacteriaceae family in older individuals, and this has been associated with an increase in inflammatory markers such as IL-6 or IL-8.34 Although the age threshold to define an individuals as elderly has been set at 70 years old, it has been proven that a healthy gut microbiota is affected by physiological and behavioural factors associated with aging after 74 years.35,36 Our study therefore revealed that the composition of the gut microbiota in 74 year-old patients with a lower CCQ was similar to those of the 74 year-old patients with a higher CCQ. Although we lack gut microbiota profiles for the older subjects without coronary calcium in our study, our data suggest that age is not the most important factor for the composition of the gut microbiota. This is one of the most interesting results of our work, as it is linked to the concept of vascular age,37 suggesting that the age of the cardiovascular apparatus would be associated more closely with the composition of the gut microbiota than it is with chronological age.
The main limitation of our work is that it is a pilot study with a relatively small study population and a cross-sectional design, so that it is unable to offer evidence for direct causal relationships. Nevertheless, using experimental murine models has shown that susceptibility to atherosclerosis may be transmitted by the transplantation of gut microbiota.38 Our group has also shown that the increase in genera belonging to the Enterobacteriaceae family (Escherichia/Shigella, Klebsiella, Enterobacter) which occurs in rats fed with a fat-rich diet can be reversed by administering a mitochondrial antioxidant, while several parameters associated with cardiac fibrosis also improve.39
To conclude, our work shows that there is a gut microbiota profile that is associated with the presence of coronary calcium in patients without previous CVD, and it would be interesting to validate this in studies with larger samples. Although there are no strategies to reduce the amount of coronary calcium, the gut microbiota is highly changeable due to a range of factors, chiefly diet. The possibility of preventing or even intervening CVD using nutritional strategies (probiotics, prebiotics or non-absorbable antimicrobial agents, etc.) is a highly attractive idea, although it has yet to be proven.
Conflict of interestsThe authors have no conflict of interests to declare.
This work was partially financed by a Grant from the Fundación Española de Arteriosclerosis (FEA)/Sociedad Española de Arteriosclerosis (SEA) 2016 for Basic Research, granted during the XXIX SEA National Congress held in Granada from 18 to 20 May 2016. Javier Modrego is a member of the personnel of the Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV).
The following is Supplementary data to this article:
Please cite this article as: Ortega-Madueño I, Modrego J, Gómez-Gordo R, Ortega-Hernández A, Pérez de Isla L, Muñoz JC, et al. Relación entre la cuantificación de calcio coronario y la composición de la microbiota intestinal en sujetos sin enfermedad cardiovascular previa: estudio piloto. Clin Investig Arterioscler. 2022;73:205–215.