




Es indudable la relación del cLDL y el riesgo cardiovascular, así como de los beneficios del tratamiento con estatinas. Una vez conseguido el objetivo de cLDL, son notables las evidencias que demuestran la persistencia de un elevado riesgo cardiovascular, concepto denominado riesgo residual.
El riesgo residual de origen lipídico se fundamenta en la dislipidemia aterogénica, caracterizada por un aumento de triglicéridos y de las lipoproteínas ricas en triglicéridos, un descenso del cHDL y alteraciones cualitativas de las partículas LDL. Las medidas más utilizadas para identificar esta dislipidemia se basan en la determinación de colesterol total, triglicéridos, HDL, colesterol no HDL y colesterol remanente, además de las apolipoproteínas B100 y la lipoproteína(a) en determinados casos.
El tratamiento de la dislipidemia aterogénica se basa en la pérdida de peso y ejercicio físico. En cuanto al tratamiento farmacológico, no tenemos evidencia del beneficio cardiovascular con los fármacos dirigidos al descenso de triglicéridos y cHDL; el fenofibrato parece tener eficacia en situaciones de dislipidemia aterogénica.
There is no doubt about the relationship between LDL-c and cardiovascular risk, as well as about the benefits of statin treatment. Once the objective of LDL-c has been achieved, the evidences that demonstrate the persistence of a high cardiovascular risk, a concept called residual risk, are notable.
The residual risk of lipid origin is based on atherogenic dyslipidemia, characterized by an increase in triglycerides and triglyceride-rich lipoproteins, a decrease in HDL-c and qualitative alterations in LDL particles. The most commonly used measures to identify this dyslipidemia are based on the determination of total cholesterol, triglycerides, HDL, non-HDL cholesterol and remaining cholesterol, as well as apolipoprotein B100 and lipoprotein (a) in certain cases.
The treatment of atherogenic dyslipidemia is based on weight loss and physical exercise. Regarding pharmacological treatment, we have no evidence of cardiovascular benefit with drugs aimed at lowering triglycerides and HDL-c, fenofibrate seems to be effective in situations of atherogenic dyslipidemia.
Actualmente se considera prioritario para la reducción del riesgo cardiovascular (RCV) el descenso del colesterol unido a las lipoproteínas de baja densidad (cLDL). Este criterio está basado en una amplia evidencia epidemiológica y en estudios prospectivos de intervención tanto en prevención primaria como secundaria que demuestran que una reducción del cLDL entre el 25 y el 40% se asocia a un descenso del RCV del 9 al 38%, en función del nivel inicial de ese riesgo1-11. Varios metaanálisis basados en estudios epidemiológicos prospectivos han demostrado una relación lineal continua entre las concentraciones de cLDL y el riesgo de enfermedad vascular arteriosclerótica. En The Emerging Risk Factors Collaboration, con los datos de 302.430 personas, sin enfermedad cardiovascular (ECV) en el momento de la inclusión, procedentes de 68 estudios prospectivos, se comprobó que la concentración plasmática de cLDL se asoció de forma lineal al aumento de riesgo de infarto de miocardio o muerte de causa cardiovascular12. De forma similar, en el Prospective Studies Collaboration, en un metaanálisis de 892.337 personas sin ECV en la incorporación al estudio, procedentes de 61 estudios prospectivos de cohortes, se confirmó una fuerte asociación lineal entre la colesterolemia y el riesgo de mortalidad por cardiopatía isquémica13.
El colesterol LDL como parte fundamental del riesgo cardiovascularEl tratamiento con estatinas ha demostrado una clara eficacia en la prevención tanto primaria como secundaria de la ECV, al ser los fármacos de elección para la reducción del cLDL. En el Cholesterol Treatment Trialists’ (CTT) Collaboration, en un metaanálisis de 26 estudios de intervención con estatinas con casi 170.000 individuos, el tratamiento con estatinas se asoció con una reducción lineal de episodios cardiovasculares14. En este metaanálisis, la reducción absoluta de cLDL fue directamente proporcional a la reducción de los principales episodios vasculares, con mayor beneficio cuanto más acentuada era la reducción de cLDL. Por cada reducción de cLDL de 1 mmol/L (39mg/dl) se redujo el riesgo de episodios vasculares en un 20% aproximadamente, de manera independiente a la concentración inicial de colesterol, lo que implica que una reducción adicional de 2-3 mmol/L disminuiría el riesgo en aproximadamente 40-50%. Estos hallazgos pusieron de manifiesto que el objetivo principal para los pacientes con alto RCV debe ser lograr la mayor reducción posible del cLDL sin aumentar el riesgo de miopatía, con independencia de las cifras objetivo propuestas en ese momento. Los datos de este metaanálisis fueron confirmados por el estudio SEARCH, que comparaba la utilización de 80mg/día vs. 20mg/día de simvastatina en más de 12.000 pacientes en prevención secundaria seguidos durante 7 años: se corroboró que las mayores reducciones de cLDL se asociaban a mayor descenso del RCV15.
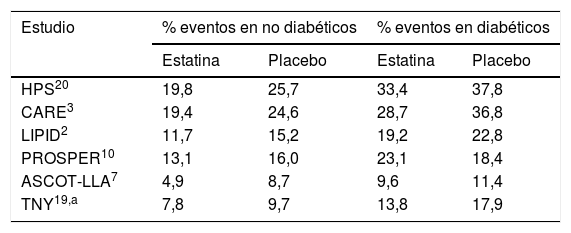
El descenso del cLDL en pacientes con diabetes mellitus de tipo 2 (DM2) es de especial importancia para reducir su elevado RCV. En el Collaborative Atorvastatin Diabetes Study (CARDS) realizado en 2.838 pacientes diabéticos sin historia cardiovascular previa, se redujo el cLDL un 40% y la incidencia de episodios cardiovasculares un 37%, en un seguimiento de hasta 9 años16. El estudio CTT confirmó el beneficio de la disminución del cLDL con estatinas en pacientes diabéticos, con un descenso del 21% de los episodios cardiovasculares por cada mmol/L de reducción del cLDL17. Un subgrupo de casi 5.000 pacientes diabéticos del estudio IMPROVE-IT confirmó que la asociación de simvastatina y ezetimiba, además de conseguir mayor descenso del cLDL (0,4 mmol/L), reducía en un 14% los episodios cardiovasculares18. Sin embargo, cabe destacar que, a pesar de los beneficios cardiovasculares de utilizar dosis máximas de estatinas19, en los pacientes con DM2 el RCV permanece elevado (tabla 1) y su expectativa de vida es más reducida11,20.
Diferentes estudios confirman que en los pacientes con DM2 permanece un elevado riesgo cardiovascular residual tras el tratamiento con estatinas
| Estudio | % eventos en no diabéticos | % eventos en diabéticos | ||
|---|---|---|---|---|
| Estatina | Placebo | Estatina | Placebo | |
| HPS20 | 19,8 | 25,7 | 33,4 | 37,8 |
| CARE3 | 19,4 | 24,6 | 28,7 | 36,8 |
| LIPID2 | 11,7 | 15,2 | 19,2 | 22,8 |
| PROSPER10 | 13,1 | 16,0 | 23,1 | 18,4 |
| ASCOT-LLA7 | 4,9 | 8,7 | 9,6 | 11,4 |
| TNY19,a | 7,8 | 9,7 | 13,8 | 17,9 |
En todos los estudios se evaluó muerte cardiovascular e infarto de miocardio no fatal.
En un reciente metaanálisis diseñado para evaluar la prevención de episodios vasculares graves con diferentes tratamientos hipolipidemiantes, realizado sobre 19 ensayos clínicos y 152.507 pacientes en prevención secundaria y con un seguimiento medio de 4 años21, se comprobó que la reducción del riesgo relativo era más importante cuanto mayor era la concentración de cLDL inicial o mayor era su reducción respecto a ella. De hecho, cada mmol/L de reducción de cLDL daba lugar a una reducción del riesgo relativo del 19% de los episodios cardiovasculares graves. Las 3conclusiones más importantes de este metaanálisis son las siguientes: el tratamiento más intensivo mostró una reducción significativa de muerte (cardiovascular y de cualquier causa), la reducción de episodios cardiovasculares es proporcional a la reducción del cLDL y la intensificación del tratamiento con fármacos no estatinas se asocia con reducciones significativas de morbilidad en población de muy alto riesgo.
El nuevo tratamiento hipolipidemiante basado en los inhibidores de la proproteína convertasa subtilisina-kexina tipo 9 (iPCSK9), usados tanto en monoterapia como asociados a estatinas, reducen la concentración de cLDL en un 45-60% aproximadamente. El estudio FOURIER abarcó a 27.564 pacientes con ECV y niveles de cLDL > 70mg/dl que seguían terapia con estatinas y que fueron aleatorizados a evoloculmab o placebo, durante 48 semanas22. La rama del fármaco en estudio redujo el cLDL en un 59%, con una diferencia absoluta de 53,4mg/dl respecto al grupo placebo. Tras una media de 2,2 años de seguimiento, redujo el objetivo primario (muerte cardiovascular, infarto de miocardio, accidente vascular cerebral, revascularización u hospitalización por angina inestable) en un 15% y se redujo el riesgo en un 11% por cada 38,6mg/dl de descenso del cLDL. El estudio ODISSEY OUTCOMES23 incluyó a 18.924 pacientes de 57 países que presentaron un infarto de miocardio o angina inestable en los 12 meses previos. Los pacientes se seleccionaron por uno de los siguientes criterios: cLDL ≥ 70mg/dl, colesterol no HDL ≥ 100mg/dl o apolipoproteína (Apo) B ≥ 80mg/dl. Después del tratamiento intensivo durante 2-16 semanas con estatinas, fueron aleatorizados para recibir alirocumab o placebo. Después de un seguimiento de entre 2y 5años (media de 2,8 años), los resultados recientemente publicados24 demostraron que los niveles de cLDL fueron de 53,3mg/dl en el grupo alirocumab comparados con 101,4mg/dl en el grupo placebo (reducción absoluta del 54,7%). El endpoint primario de eventos cardiovasculares mayores fue significativamente menor en el grupo alirocumab que en el grupo placebo (9,5 vs. 11,1%). Concretamente con el fármaco en estudio, el infarto de miocardio no fatal se redujo en un 14%, el accidente vascular cerebral en un 27% y la angina inestable en un 39%. En el análisis post hoc por nivel de cLDL al inicio del estudio, los pacientes con un cLDL ≥ 100mg/dl experimentaron mayores reducciones en todos los criterios de valoración.
Desconocemos en gran medida el efecto de los iPCSK9 sobre el resto de las fracciones lipídicas con potencial aterogénico que podrían influir en el RCV; sin embargo, disponemos de un reciente metaanálisis con los datos agrupados de 10 estudios en fase 3pertenecientes al ODISSEY. En él se analizó el efecto de alirocumab en pacientes con síndrome metabólico: el descenso de Apo B en estos pacientes fue del 35 al 55% y el del colesterol no ligado a lipoproteínas de alta densidad (c-no-HDL) del 38 al 54%, porcentajes similares a los presentados en los pacientes sin síndrome metabólico25. En otro metaanálisis reciente con evolocumab26 en pacientes con DM2, el descenso del c-no-HDL fue del 55% vs. ezetimiba y del 34% vs. placebo. En el estudio ODISSEY LONG TERM27 el descenso del c-no-HDL fue del 51,6 vs. el 0,7% del placebo y la Apo B descendió en un 52,8% vs. 1,2% con el placebo. Por otra parte, está el estudio ODISSEY DM-DYSLIPIDEMIA realizado en pacientes con DM2 y dislipidemia (c-no-HDL ≥ 100mg/dl y triglicéridos [TG] ≥ 100mg/dl), que fueron aleatorizados para recibir alirocumab o, además del tratamiento con la dosis máxima tolerada de estatina, otros hipolipidemiantes según uso habitual (fenofibrato, ezetimibe, omega-3, ácido nicotínico o solo dieta). En este estudio, alirocumab redujo el cLDL (43%), Apo B (32,3%) y el número de partículas LDL (37,8%) vs. el tratamiento usual elegido (p <0,0001)28. Según estos datos, podemos concluir que los iPCSK9 tienen una clara y significativa acción sobre el conjunto de las lipoproteínas aterogénicas.
Riesgo residual no dependiente de colesterol LDLCon los datos referidos, queda absolutamente clarificada la influencia del cLDL en el RCV y que, cuanto más intenso sea el descenso de esta lipoproteína, mayor será el descenso del RCV. No obstante, a pesar de obtener cifras de cLDL en el rango de los objetivos marcados por diferentes sociedades científicas e incluso de conseguir las cifras más bajas posibles tanto con estatinas como con fármacos no estatinas, persiste un elevado, inaceptable y comprobado RCV y, de hecho, persiste un elevado porcentaje de episodios cardiovasculares, especialmente en pacientes en prevención secundaria. Este RCV, persistente a pesar del tratamiento, es debido en gran medida a otros factores lipídicos diferentes del cLDL pero con potencial acción aterogénica29, por lo que es denominado RCV residual de origen lipídico.
Este riesgo residual de origen lipídico refleja aspectos relacionados con las lipoproteínas ricas en TG (LRTG) que comprende sobre todo a las de muy baja densidad (VLDL). La hidrólisis enzimática de las VLDL da lugar a la formación de partículas más pequeñas y más densas, enriquecidas en colesterol. Estudios experimentales indican que estas lipoproteínas podrían contribuir al desarrollo de la ECV30,31, concepto que es asumido por diferentes consensos y sociedades científicas32,33.
Por otra parte, algunos estudios epidemiológicos han puesto de manifiesto la importancia del riesgo residual de origen lipídico en el RCV total. En el Residual Risk Lipids and Standard Therapies (REALIST), se ha comprobado que los TG y el cHDL están estrechamente relacionados con el riesgo residual en pacientes con síndrome coronario agudo y con cLDL entre 70 y 130mg/dl; además, las concentraciones más elevadas de TG asociadas a las más bajas de cHDL multiplican por 10el riesgo de enfermedad cardiaca coronaria. En el Prospective Cardiovascular Münster, el RCV para un infarto agudo de miocardio se multiplicó por 5en pacientes con cifras de cLDL en el rango establecido, pero con aumento de TG o descenso del cHDL34.
A la vista de estos resultados, es importante la identificación de otros factores lipídicos diferentes al cLDL asociados al riesgo residual de origen lipídico, no solo para su corrección sino también para su identificación en los enfermos con un elevado RCV, que frecuentemente presentan estas alteraciones lipídicas, como ocurre por ejemplo en las situaciones de resistencia a la insulina tales como la DM2, obesidad o síndrome metabólico.
La acumulación de pruebas ha respaldado la importancia de la aterogenicidad de las LRTG y sus remanentes, a menudo en combinación con baja concentración de cHDL, incluso son considerados actualmente como objetivos terapéuticos secundarios en los pacientes de alto/muy alto RCV. La Iniciativa de Reducción de Riesgo Residual (R3i) ha destacado la importancia de esta dislipidemia como contribuyente al RCV residual relacionado con los lípidos35. De hecho, un análisis post hoc de ensayos con fibratos mostró que las personas con este perfil dislipidémico tuvieron una reducción relativa del 35% en el riesgo de episodios cardiovasculares, frente al 6% en aquellos individuos sin esta dislipidemia36.
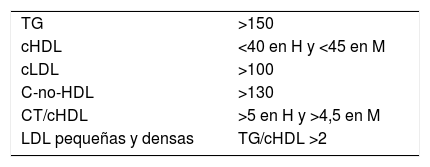
De la asociación del aumento de las LRTG, descenso de cHDL y el incremento de partículas de LDL pequeñas y densas, nace el concepto de la dislipidemia aterogénica (DA), responsable en gran medida del riesgo residual de origen lipídico (tabla 2). Si la prevalencia del síndrome metabólico es aproximadamente del 22 al 39% de la población adulta y los componentes lipídicos de dicho síndrome coinciden con los de DA, podríamos estimar la gran prevalencia de este trastorno lipídico en la población general. Analizaremos a continuación los componentes de la DA y las medidas necesarias para su diagnóstico.
Actualmente existe suficiente evidencia científica para considerar a la hipertrigliceridemia como un factor de riesgo independiente para la ECV37. Un metaanálisis que analizó la asociación entre TG y ECV en población general, basado en estudios prospectivos con un total de 65.863 hombres y 11.089 mujeres, evidenció que la elevación de 89mg/dl en la concentración de TG estaba asociada con un incremento del RCV del 12% en los varones y del 37% en las mujeres, después de ajustar por colesterol total, cLDL, cHDL, índice de masa corporal, presión arterial y diabetes38.
La elevación de los TG se asocia de forma independiente a la DM2, al síndrome metabólico, a la obesidad y a la enfermedad renal crónica39; además, este incremento se ve influido por la dieta, algunos fármacos, factores genéticos y, muy especialmente, por el estilo de vida. Se estima que hasta una cuarta parte de la población adulta en EE. UU. presenta hipertrigliceridemia, definida esta como TG>150mg/dl40. La hipertrigliceridemia grave, definida por la National Lipid Association guidelines como TG>500mg/dl, es un claro factor de riesgo para la pancreatitis aguda41; sin embargo, se ha demostrado que los TG moderadamente elevados se asocian de forma independiente con un mayor riesgo de ECV, incluso en pacientes tratados de forma eficaz con estatinas para reducir el cLDL42,43.
El estudio Emerging Risk Factors Collaboration, en población general sin ECV previa, puso de manifiesto una asociación de la trigliceridemia con distintos episodios cardiovasculares, aunque dicha asociación desaparecía cuando se ajustaba por cHDL y c-no-HDL44. En el Copenhagen General Population Study, se observó también una potente asociación entre los TG posprandiales y el RCV45. En el seguimiento del estudio Bezafibrate Infarction Prevention se analizaron los datos de mortalidad tras un periodo de más de 20años de seguimiento y en más de 15.000 pacientes en prevención secundaria, y se comprobó que el riesgo de mortalidad a 22 años se incrementó en un 68% en pacientes con hipertrigliceridemia en comparación con los pacientes con TG bajos o normales (p <0,001). Por tanto, los TG elevados en pacientes con ECV están asociados con un mayor riesgo de mortalidad a largo plazo, que es independiente del cHDL. La hipertrigliceridemia grave (TG>500mg/dl) identifica una población con un riesgo de mortalidad especialmente aumentado46.
En el estudio PROVE-IT TIMI, realizado en pacientes con síndrome coronario agudo en tratamiento con estatinas y cLDL en objetivo (<70mg/dl), una concentración de TG>150mg/dl se asociaba de forma independiente con una menor tasa de recurrencias40. El análisis agrupado de los estudios IDEAL y TNT demostró, asimismo, que, en pacientes en prevención secundaria tratados con estatinas, el descenso de TG se acompañaba de una reducción en la incidencia de episodios cardiovasculares, aunque esta asociación se atenuaba al ajustar por cHDL47.
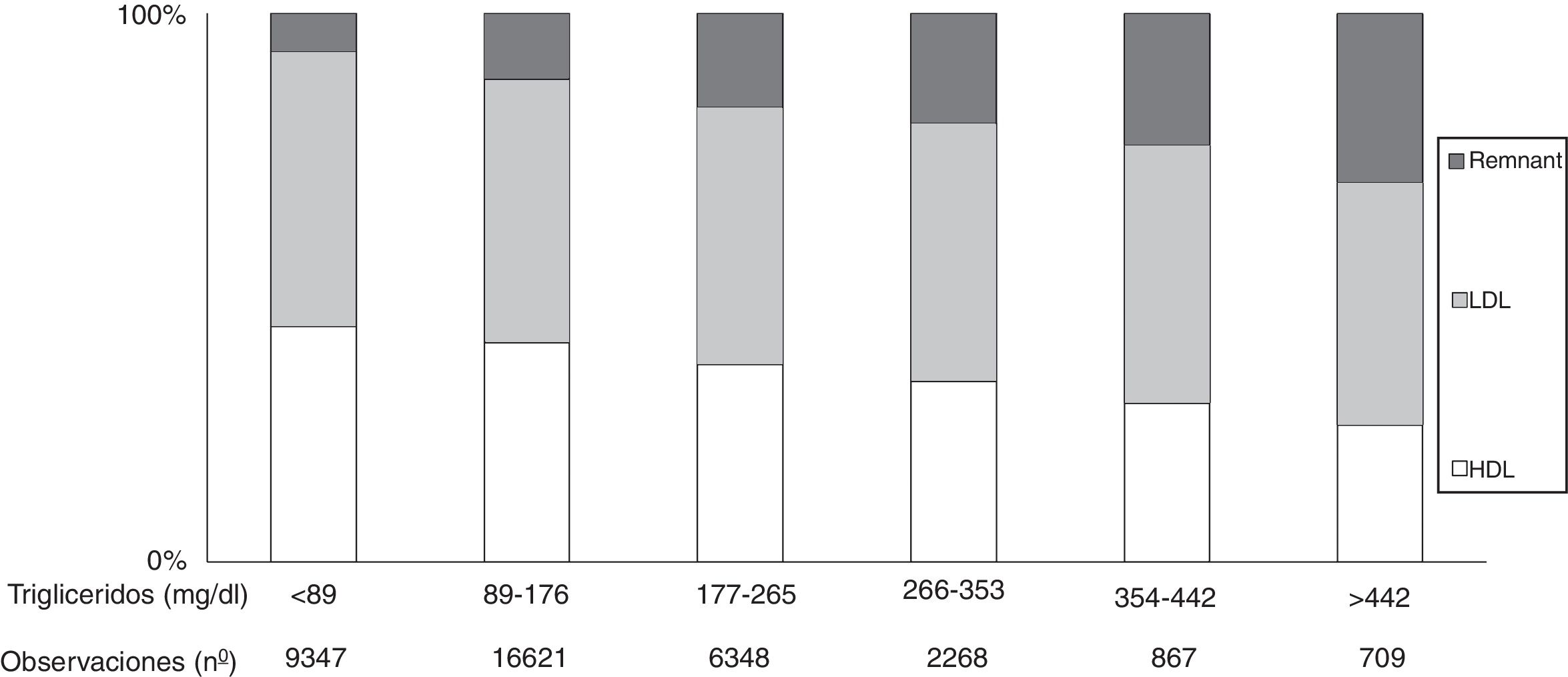
Es conocido que los niveles de TG plasmáticos se corresponden con los niveles de LRTG y sus remanentes (fig. 1)32. Sin embargo, la hipertrigliceridemia también suele ir acompañada de otras alteraciones lipoproteicas, como el aumento de las VLDL y la Apo C-III, predominio de partículas LDL pequeñas y densas y niveles reducidos de cHDL, todas ellas en estrecha relación con un mayor riesgo de ECV48.
, mientras que las LDL no presentan modificaciones y las HDL tendían a disminuir. En estas condiciones, el colesterol remanente representa el colesterol total transportado en restos de IDL, VLDL y quilomicrones. Fuente: Modificado de Chapman et al.32.")
Colesterol en lipoproteínas en función del aumento de los niveles de triglicéridos en la población general. Basado en el estudio de población general de Copenhague. El 9% de los hombres y el 6% de las mujeres tomaban estatinas. A medida que se incrementan los triglicéridos se incrementan las lipoproteínas remanentes (VLDL, IDL y remanente), mientras que las LDL no presentan modificaciones y las HDL tendían a disminuir. En estas condiciones, el colesterol remanente representa el colesterol total transportado en restos de IDL, VLDL y quilomicrones. Fuente: Modificado de Chapman et al.32.
El hecho de que los resultados de los estudios con fármacos reductores de TG hayan producido resultados cardiovasculares inconsistentes49 explica que aún exista cierta incertidumbre con respecto al papel causal directo de las LRTG en la progresión del proceso arteriosclerótico y la ECV.
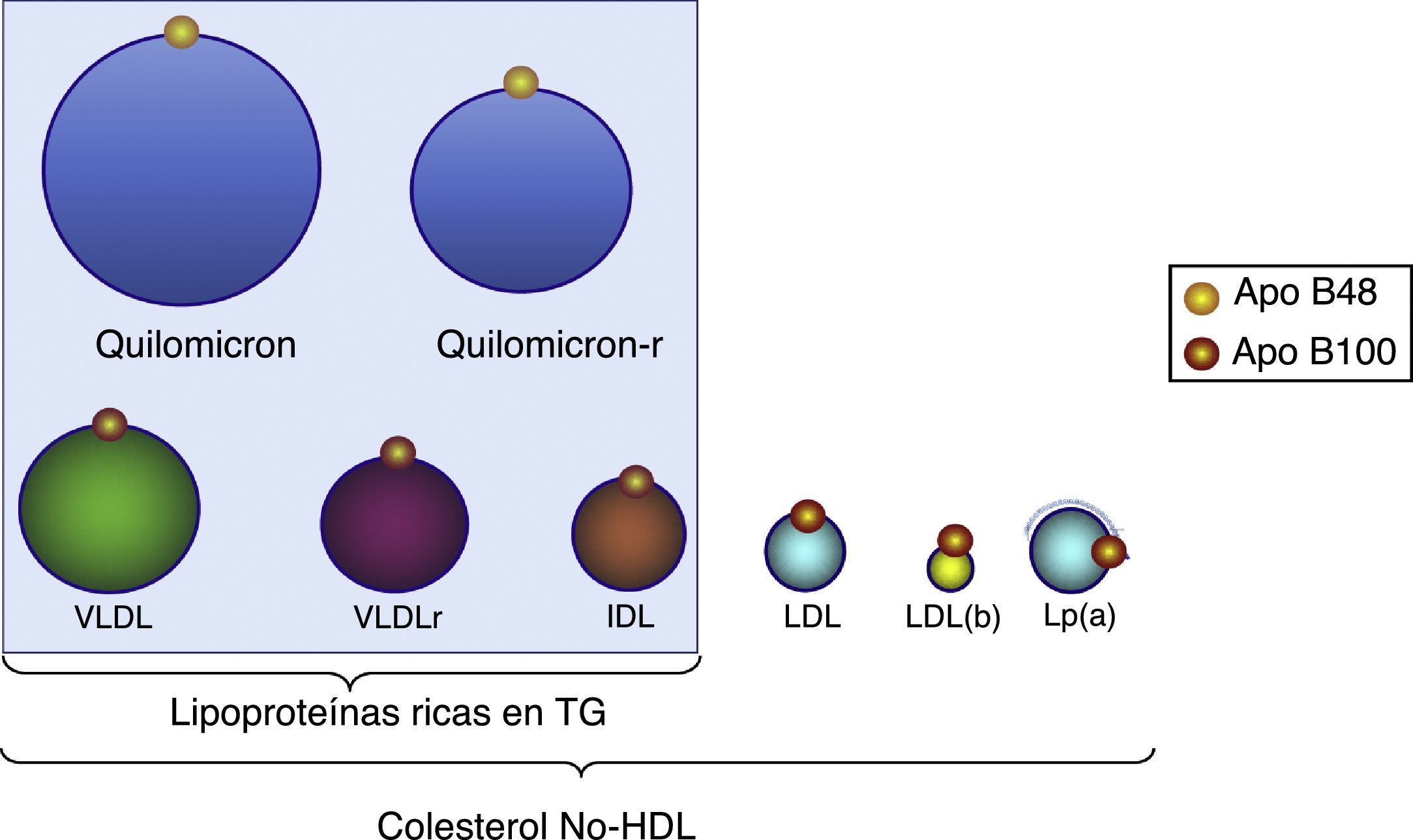
Lipoproteínas ricas en triglicéridosLos TG son transportados en el plasma por las VLDL, quilomicrones y los restos de estos originados durante su metabolismo (remanentes). Las LRTG son las partículas de lipoproteínas más grandes y se caracterizan por las Apo que contienen, como la Apo B100, que se asocia con VLDL e IDL (fig. 2) y la Apo B48 que se asocia con los quilomicrones y sus remanentes.
: LDL pequeña y densa; quilomicron-r: quilomicron residual o remnant; VLDLr: VLDL residual o r.")
Distribución de las lipoproteínas ricas en triglicéridos y partículas que abarca el c-no-HDL. Todas estas lipoproteínas contienen una molécula de Apo B100, por lo que la medida de esta apoproteína indica el número de partículas con potencial aterogénico. LDL(b): LDL pequeña y densa; quilomicron-r: quilomicron residual o remnant; VLDLr: VLDL residual o r.
Las LRTG son muy heterogéneas por su diferente tamaño, densidad y composición. Están compuestas por un núcleo neutro de TG y ésteres de colesterol y una monocapa superficial que comprende fosfolípidos, colesterol libre y las Apo que participan en la regulación del transporte y su metabolismo50.
Se ha propuesto que es principalmente el contenido de colesterol de las LRTG el que contribuye de forma directa a la progresión de la aterosclerosis, en lugar de los propios TG51. Al igual que las LDL, las LRTG empobrecidas en TG y enriquecidas en colesterol pueden penetrar la íntima arterial y ser atrapadas en el espacio subendotelial, donde son fagocitadas por los macrófagos, lo que contribuye a la formación de células espumosas y a la formación y progresión de la placa de ateroma. Se acepta que las LRTG son igual o incluso más aterogénicas que las LDL ya que, a diferencia de estas, las partículas remanentes de LRTG pueden ser absorbidas directamente por los macrófagos sin modificación oxidativa; además, por su mayor tamaño, transportan más colesterol por partícula que las LDL52. También se ha demostrado que los remanentes de LRTG promueven la disfunción endotelial, lo que potencia la aterogénesis53.
La hidrólisis de las LRTG mediada por la lipoproteinlipasa da como resultado una alta concentración de ácidos grasos libres en el endotelio o incluso dentro de la íntima arterial. Estos productos lipolíticos, junto con las propias LRTG, activan varias vías de señalización proinflamatorias, procoagulantes y proapoptóticas, que desempeñan un papel fundamental en la patogénesis de la aterosclerosis. Conocemos que los ácidos grasos libres oxidados aumentan la expresión de interleucinas inflamatorias y citocinas, que conducen a la inflamación endotelial54, mientras que los remanentes de las LRTG facilitan la expresión endotelial de las moléculas de adhesión55, que son proaterogénicas, al facilitar la migración transendotelial de leucocitos, además de la activación de monocitos y neutrófilos56.
Por otra parte, las LRTG remanentes aumentan la producción de las especies reactivas de oxígeno con el consiguiente incremento de la permeabilidad endotelial, promueven la adhesión de leucocitos e incrementan la secreción de citocinas proapoptóticas, factor de necrosis tumoral α e interleucina 1β, lo que conduce a la lesión vascular y al desarrollo del proceso arteriosclerótico57.
Las LRTG y sus partículas remanentes aumentan la agregación plaquetaria y amplifican la cascada de la coagulación a través del complejo protrombinasa y el incremento de la expresión del inhibidor-1 del activador del plasminógeno, además de favorecer la expresión endotelial del factor tisular58,59. Finalmente, existen pruebas de que las LRTG remanentes suprimen los efectos ateroprotectores y antiinflamatorios de HDL60 y se ha demostrado que se correlacionan significativamente con el deterioro de la vasodilatación coronaria61. Los estudios de aleatorización mendeliana en los que se han utilizado los genes implicados en las vías del metabolismo de las LRTG (Apo A5, Apo C3) han demostrado que estas lipoproteínas son causales de ECV.
Colesterol remanenteEl colesterol residual o remanente corresponde al colesterol contenido en las LRTG, es decir, en las VLDL, en las lipoproteínas de densidad intermedia (IDL) y en los quilomicrones remanentes. Puede ser calculado de forma simple sustrayendo al colesterol total el cLDL y el cHDL. La diferencia con el c-no-HDL es que este contiene colesterol residual más cLDL. La importancia del colesterol remanente radica en identificarlo en sujetos con cLDL normal o bajo, ya que en estos casos se pueden presentar valores elevados que no se identificarían con la sola valoración del c-no-HDL. Actualmente se considera al colesterol remanente un importante factor de RCV51,62.
Existen datos que demuestran la asociación entre altas concentraciones de colesterol remanente y el riesgo elevado de cardiopatía isquémica63, infarto de miocardio64 y mortalidad por todas las causas65. En el recientemente publicado The Copenhagen General Population Study realizado sobre un total de 106.216 pacientes seguidos durante 11 años, estos fueron distribuidos por cuartiles de colesterol remanente y en función de que presentaran bajo peso, peso normal, sobrepeso u obesidad. La conclusión de este estudio es que el colesterol remanente y el índice de masa corporal se correlacionaron positivamente y que es un factor de riesgo para el infarto de miocardio independiente del parámetro antropométrico66. En un resultado preliminar del mismo estudio, realizado sobre 90.000 sujetos en población general, se comprobó que el incremento progresivo del colesterol remanente y el cLDL se asociaron con el incremento del riesgo de cardiopatía isquémica e infarto de miocardio; sin embargo, aunque las concentraciones crecientes de colesterol remanente se asociaron a una mayor mortalidad por todas las causas, no ocurrió lo mismo con el cLDL. Incluso después de reducir el cLDL a las concentraciones recomendadas, todavía existía un riesgo sustancial de ECV, por lo que concluyen los autores la necesidad, para los futuros estudios de intervención, de reducir tanto el cLDL como el remanente67.
Colesterol no HDLEl c-no-HDL equivale a la suma del colesterol de las LDL, de la lipoproteína(a) (Lp[a]), de las VLDL y de las IDL. Su cálculo se basa simplemente en sustraer al colesterol total el cHDL. En la actualidad se considera un marcador fiable de RCV, al englobar todas las lipoproteínas aterogénicas que contienen colesterol y Apo B; por otra parte, es fácil de calcular y no tiene interferencias con el resto de las determinaciones lipídicas. Es una medida de gran utilidad en situaciones clínicas relacionadas con la resistencia a la insulina, tan frecuentes como la DM2, el síndrome metabólico o la obesidad visceral, en las que la DA es un trastorno característico, por lo que puede considerarse un importante marcador del riesgo residual de origen lipídico. En estos pacientes se ha recomendado que el objetivo terapéutico más preciso sea el c-no-HDL, o bien la Apo B, puesto que ambos parámetros se correlacionan mejor con el RCV que el cLDL68, incluso existen evidencias de que el c-no-HDL es superior al cLDL en la evaluación del RCV69.
La situación descrita confirma que, aunque el cLDL pueda permanecer más o menos estable, a medida que aumentan las lipoproteínas aterogénicas restantes (lipoproteínas que contienen Apo B) y disminuye el cHDL, el riesgo se incrementa, lo que representa todo el colesterol aterogénico y no solo al cLDL. Si en esta situación se empleara solo el cLDL, estaríamos infraestimando el riesgo porque no reflejaría el auténtico aumento de lipoproteínas aterogénicas70. En un reciente estudio realizado en pacientes con DM2 y con cifras de cLDL < 100mg/dl, se ha comprobado que la relación Apo B/Apo A-I elevada (>0,57) se asocia de forma independiente al riesgo de arteriosclerosis carotídea71.
En las últimas guías clínicas para de la Asociación Americana de Endocrinólogos Clínicos y el Colegio Americano de Endocrinólogos, se explicita el aumento del c-no-HDL como factor mayor de riesgo con igual importancia que el cLDL o el colesterol total y equivalente a la hipertensión arterial, DM2, enfermedad renal crónica, tabaquismo o antecedentes familiares de ECV. Esta misma guía establece 5categorías de riesgo (bajo, moderado, alto, muy alto y extremo) y fija objetivos terapéuticos para el c-no-HDL para cada una de las categorías de riesgo mencionadas72. También se recomienda el cálculo del c-no-HDL para la estratificación del riesgo en individuos con TG moderadamente elevados (>200mg/dl), diabetes y en prevención secundaria. Además, en sujetos con sospecha de resistencia a la insulina (síndrome metabólico) el c-no-HDL debe ser evaluado para considerar la carga de lipoproteínas aterogénicas.
Un metaanálisis de 8 estudios con 62.154 pacientes tratados con estatinas demostró que el c-no-HDL tenía una mejor correlación con el RCV que el propio cLDL y la Apo B; incluso, los sujetos que alcanzaban solo los objetivos terapéuticos de cLDL mostraban un incremento de riesgo del 32% (IC 95%: 1,17-1,50) en comparación con aquellos que conseguían ambos objetivos71. En otro reciente metaanálisis realizado con 9estudios prospectivos en población general, que abarcaban a 448.732 individuos, se demostró que los pacientes con mayor nivel de c-no-HDL al inicio del estudio presentaron un mayor riesgo de ECV (RR 1,79; IC 95%: 1,68-1,91) comparados con los que presentaron el nivel más bajo73.
Número de partículas LDLLas subfracciones de las partículas LDL pueden ser determinadas por diferentes métodos: gradiente de densidad por ultracentrifugación, electroforesis, cromatografía, aunque la utilización de la espectroscospia por resonancia nuclear magnética es actualmente el método más eficiente, ya que proporciona una cuantificación rápida del tamaño y concentración de las partículas de LDL y varias subclases de lipoproteínas, incluidas VLDL, IDL, LDL y HDL, por lo que es una potencial e importante ayuda para evaluar rigurosamente el RCV en la clínica práctica.
Grandes estudios prospectivos (EPIC-Norfolk, Framingham Offsping, MESA, Women's Health Studies) han demostrado que la cuantificación de partículas LDL tiene mayor valor predictivo de RCV que el propio valor de cLDL. Este concepto es importante, ya que muchos pacientes con DA (DM2, síndrome metabólico) tienen incremento del número de partículas LDL sin presentar incremento del cLDL. En esta situación, los pacientes a menudo tienen concentraciones más altas de lipoproteínas ricas en TG, lo que puede explicar el mayor riesgo de ECV. También el número de partículas LDL puede considerarse un marcador de ECV subclínica, al asociarse al espesor íntima-media y al calcio arterial en mayor medida que el cLDL74.
La presencia de subclases de LDL depende de la disponibilidad hepática de los TG: si es baja, el hígado segrega principalmente VLDL1 (rica en TG) e IDL. Las VLDL1 son modificadas por la lipoproteinlipasa y la lipasa hepática para producir LDL-III, que por medio de la propia lipasa hepática y la proteína transferidora de ésteres de colesterol (PTEC) acaba formando LDL pequeñas y densas. A partir de las IDL, a través de la lipoproteinlipasa se formarán LDL-I. Por el contrario, cuando la disponibilidad de TG es elevada, se produce un patrón distinto de secreción: se producen VLDL1 grandes y VLDL2, después de la acción de la lipoproteinlipasa y la lipasa hepática, las primeras se transforman en LDL-IV y las segundas en IDL1 y en LDL-II, por ambas vías y por medio de la lipasa hepática y la PTEC se producen LDL pequeñas y densas75. En resumen, podríamos afirmar que el aumento de las LRTG produce un incremento del intercambio de estos con ésteres de colesterol provenientes de las diferentes partículas LDL mediante la acción de la PTEC y, por medio de esta acción enzimática, se incrementa la formación de partículas LDL más pequeñas y densas76.
Las partículas de LDL pequeñas y densas tienen una menor afinidad por el receptor LDL, lo que retrasa su aclaramiento de la circulación. Esta mayor permanencia en plasma, junto con su menor tamaño, facilita la penetración en la íntima arterial y su retención en el espacio subendotelial por su mayor afinidad con los proteoglicanos. Además, estas partículas son más susceptibles a la oxidación, que es determinante en el proceso de aterogénesis. Este patrón de partículas LDL está claramente incrementado en la DM2 y está relacionado de forma estrecha con la concentración de LRTG, ya que se observa hasta en un 90% de los pacientes con TG>150mg/dl.
En la mayoría de las ocasiones, la concentración de cLDL y el número de las partículas de LDL están estrechamente relacionados, por lo que el cLDL plasmático es un buen sustituto para la concentración de partículas LDL. Sin embargo, en ciertas condiciones (síndrome metabólico, DM2 e hipertrigliceridemia), la concentración de cLDL y el número de partículas LDL puede ser discordante, debido al predominio de las partículas LDL pequeñas y densas y, por lo tanto, el cLDL plasmático puede no reflejar la concentración de partículas de LDL. Bajo estas condiciones, la medición directa del número de partículas LDL o la concentración de Apo B (reconociendo que cada partícula de LDL contiene una sola molécula de Apo B) puede reflejar con mayor efecto causal que el propio LDL para la ECV arteriosclerótica77.
La cantidad de estas partículas puede ser estimada de forma indirecta mediante el cociente TG/cHDL, que si es superior a 2 indica su incremento y el rango para ser considerado en la DA78.
Actualmente está aceptado que todas las partículas LDL deben ser consideradas aterogénicas79, hecho evidenciado por los pacientes con hipercolesterolemia familiar, que muestran un predominio de partículas de LDL grandes y una aterosclerosis prematura80. Por otra parte, una revisión de 24 estudios que informaron de las relaciones entre las subfracciones LDL y eventos cardiovasculares llegó a la conclusión de que el mayor número de partículas LDL, más que el tamaño de estas partículas, se asoció a un mayor riesgo de eventos cardiovasculares81.
HDLLas HDL son las encargadas del transporte reverso del colesterol, es decir, desde los tejidos periféricos hasta el hígado, uno de los principales mecanismos antiaterogénicos.
Las partículas de HDL son plurimoleculares, compuestas predominantemente de lípidos polares, además de proteínas (enzimas) y pequeñas cantidades de lípidos no polares, de las que existen en múltiples isoformas. Como consecuencia de su composición, son altamente heterogéneas en sus propiedades estructurales, químicas y biológicas. Existen 2tipos: HDL2, las menos densas con más contenido lipídico, y HDL3, más densas y ricas en proteínas. HDL2 y HDL3 se pueden fraccionar en distintas subclases con diferentes movilidades electroforéticas. Existen 2subclases de HDL2 y 3de HDL3.
El incremento de LRTG también da lugar a que por medio de la PTEC se transfieran TG a las HDL. El enriquecimiento de las HDL en TG las hace más susceptibles a la acción de la lipasa hepática, enzima que actúa sobre las partículas de HDL2 (mayor tamaño, menor densidad y contenido variable de colesterol) y las transforma en partículas más pequeñas de HDL3 (pequeñas, densas, con escaso contenido en lípidos), que son rápidamente aclaradas del plasma y se eliminan por vía renal. Se ha comprobado que los niveles de TG modulan la concentración de subclases de HDL, ya que el aumento de TG se acompaña de HDL de menor tamaño, especialmente debido al descenso de la subclase HDL2 e incremento de la HDL3. Estos cambios en las HDL van asociados al descenso de su contenido en colesterol libre y al aumento de TG. El resultado de este proceso es un descenso neto de las HDL y un incremento de las partículas LDL pequeñas y densas. Esta dislipidemia se suele acompañar de un incremento del c-no-HDL y, en consecuencia, de un aumento de Apo B, ya que todas las partículas no HDL llevan esta Apo. Las concentraciones elevadas de TG y el descenso del cHDL se acompañan de un elevado RCV, incluso en pacientes con el cLDL en concentraciones deseables82.
Las HDL, además de facilitar el transporte reverso de colesterol, también protegen directamente la pared vascular por otros mecanismos: disminuyen la adhesividad celular al endotelio con la consiguiente disminución de la migración endotelial de los monocitos, facilitan la producción de prostaciclina (vasodilatador e inhibidor de la agregación plaquetaria), interfieren la fibrinólisis y disminuyen los mediadores celulares y humorales de la inflamación arterial83.
En este sentido, el estudio Emerging Risk Factors Collaboration ya mencionado señalaba al cHDL como un factor de riesgo predictor independiente de episodios coronarios y cerebrovasculares, incluso cuando se ajustaba por el resto de los factores lipídicos y no lipídicos de riesgo. El estudio SPARCL, en pacientes con enfermedad cerebrovascular previa, mostró un mayor descenso en el riesgo de recurrencia de ictus cuando los niveles de cHDL eran más altos, independientemente de las cifras de cLDL84. Los metaanálisis del CTT tanto en prevención primaria como secundaria, y en pacientes tratados con estatinas, señalan de forma inequívoca que, al margen e independientemente de las cifras de cLDL, el RCV es inferior con cifras más elevadas de cHDL, aun en los casos de cLDL más bajo85,86. Otro estudio en pacientes con cardiopatía isquémica estable y con estatina de alta intensidad (TNT) también señaló el poder predictor de las concentraciones bajas de cHDL, aun con cLDL bien controlado (<70mg/dl)82. En pacientes con síndrome coronario agudo y tratados con estatinas (estudio MIRACL) se ha constatado que el cHDL, y no así el cLDL, era un predictor independiente del pronóstico a corto plazo14. Por último, un metaanálisis de 20 estudios que abarcaba un total de 70.939 pacientes con estatinas y 66.068 con placebo ha demostrado que las estatinas no alteraron la relación entre el nivel de cHDL y el RCV, por lo que los niveles bajos de cHDL permanecen significativa e independientemente asociados con un mayor riesgo a pesar de la terapia con estatinas87. Los pacientes tratados con estatinas tenían el mismo grado de riesgo asociado con menores niveles de cHDL que los sujetos control (incluso después del ajuste por los niveles de cLDL en tratamiento, edad, hipertensión, diabetes y consumo de tabaco)68. Estos hallazgos resaltan el RCV asociado con niveles bajos de cHDL, incluso en pacientes tratados con estatinas.
Lipoproteína(a)Esta lipoproteína se considera como un marcador adicional e independiente de RCV88,89. La Lp(a) tiene similitudes con la LDL, contiene también Apo B100 y, además, una Apo específica, la Apo (a), que es estructuralmente homóloga al plasminógeno. La concentración plasmática de Lp(a) está genéticamente determinada. La determinación sistemática del nivel plasmático de Lp(a) no está recomendada para la detección de riesgo en la población general; sin embargo, la medición de Lp(a) se debe considerar en personas con alto riesgo de ECV o un historial familiar de episodios cardiovasculares prematuros. El riesgo se considera significativo cuando la concentración de Lp(a) está por encima del percentil 80 (50mg/dl)90. Asimismo, se ha demostrado que Lp(a) proporciona una reclasificación correcta en la evaluación de riesgos88,91 y debe considerarse en pacientes en el límite entre riesgo alto y moderado.
Enfoque terapéutico del riesgo cardiovascular residualUn enfoque terapéutico adecuado sobre la DA debe tener un efecto positivo sobre el riesgo residual de origen lipídico, que persiste después de un empleo eficaz de las estatinas con objeto de disminuir los niveles de cLDL. Por consiguiente, la identificación de los factores lipídicos asociados al riesgo residual puede ser particularmente importante, no solo para su tratamiento sino también de cara a su detección en los pacientes con diferentes alteraciones en el perfil lipídico asociadas a un elevado RCV, como es el caso de los pacientes con DM2 y su característica DA en más del 50% de los casos.
Las diferentes guías clínicas incluyen el c-no-HDL o la Apo B como objetivos secundarios una vez alcanzado el objetivo principal en cLDL, o incluso en algunos casos se plantean como objetivos con el mismo nivel de importancia el cLDL y el c-no-HDL. En todos los casos es crucial la modificación del estilo de vida (dieta y ejercicio) para conseguir la reducción del riesgo residual. Además, existen estrategias farmacológicas para reducir dicho riesgo que comentaremos a continuación.
Dieta y ejercicioUna dieta rica en grasas monoinsaturadas mejora significativamente la sensibilidad a la insulina, situación que se asocia con una reducción en los niveles de TG, sobre todo en situación posprandial. El descenso de TG se evidencia cuando la grasa saturada es reemplazada por ácidos grasos poliinsaturados omega-6, o con altas dosis de ingesta de omega-3. Sin embargo, un enfoque terapéutico basado solo en los alimentos difícilmente llegará a alcanzar un efecto clínico significativo. Para conseguir este objetivo, pueden ser utilizados suplementos o alimentos enriquecidos de manera artificial con PUFA omega-3. En sujetos con hipertigliceridemia severa, incluso con presencia de quilomicrones en situación de ayuno, es apropiado reducir la cantidad total de grasa dietética tanto como sea posible (<30g/día). En estos pacientes se puede considerar el uso de TG de cadena mediana (de C6 a C12), que evitan la formación de quilomicrones ya que son directamente transportados y metabolizados en el hígado por vía portal92.
También es conocida la relación entre el metabolismo de los hidratos de carbono y los lípidos. Por esta razón, la ingesta de un alto contenido de hidratos de carbono en la dieta puede conducir al incremento de la concentración de TG93. Esta situación ocurre de manera especial con la ingesta de hidratos de carbono de rápida absorción (refinados) y elevado índice glucémico; este efecto se reduce considerablemente con el consumo de hidratos de carbonos complejos y con alto contenido en fibra, y es aplicable a pacientes con DM2 o síndrome metabólico.
La reducción de peso mejora la sensibilidad a la insulina, con lo que disminuyen los niveles de TG, que alcanzan reducciones de hasta el 30% que se mantienen mientras no se recupera el peso. La práctica del ejercicio físico regular reduce el nivel de TG con independencia de la pérdida ponderal. La ingesta de alcohol tiene un gran impacto en la trigliceridemia. Mientras que, en individuos con hipertrigliceridemia, incluso una pequeña cantidad de alcohol puede inducir una mayor elevación de las concentraciones de TG, en la población general la ingesta de alcohol solo repercute en el nivel de TG si es excesiva94.
Respecto al cHDL, la ingesta de ácidos grasos saturados lo incrementan de forma paralela al incremento del cLDL95. El consumo de grasa mono- o poliinsaturada prácticamente no tiene influencia en el cHDL. El incremento del consumo de hidratos de carbono en sustitución de grasa se asocia a un descenso del cHDL (por cada 1% de sustitución desciende 0,4mg/dl), efecto no influible por el contenido en fibra o el propio índice glucémico.
El consumo moderado de alcohol está asociado con mayores niveles de cHDL en comparación con los abstemios. La reducción del peso tiene un claro beneficio en los niveles de cHDL: aumenta 0,4mg/dl por cada kilogramo de disminución de peso. La actividad física aeróbica correspondiente a deambulación rápida de 3,5-4,5 km/día (o cualquier actividad equivalente) puede aumentar los niveles de cHDL entre 3 y 6mg/dl)96. Dejar de fumar también puede contribuir a la elevación del cHDL, siempre que se evite la ganancia de peso que suele ocurrir al suprimir el hábito tabáquico.
FármacosHasta el momento actual, los estudios clínicos realizados con diferentes fármacos dirigidos a la reducción de TG y al aumento de cHDL y, con ello, a reducir el riesgo residual de origen lipídico, han tenido escaso éxito en la reducción del RCV.
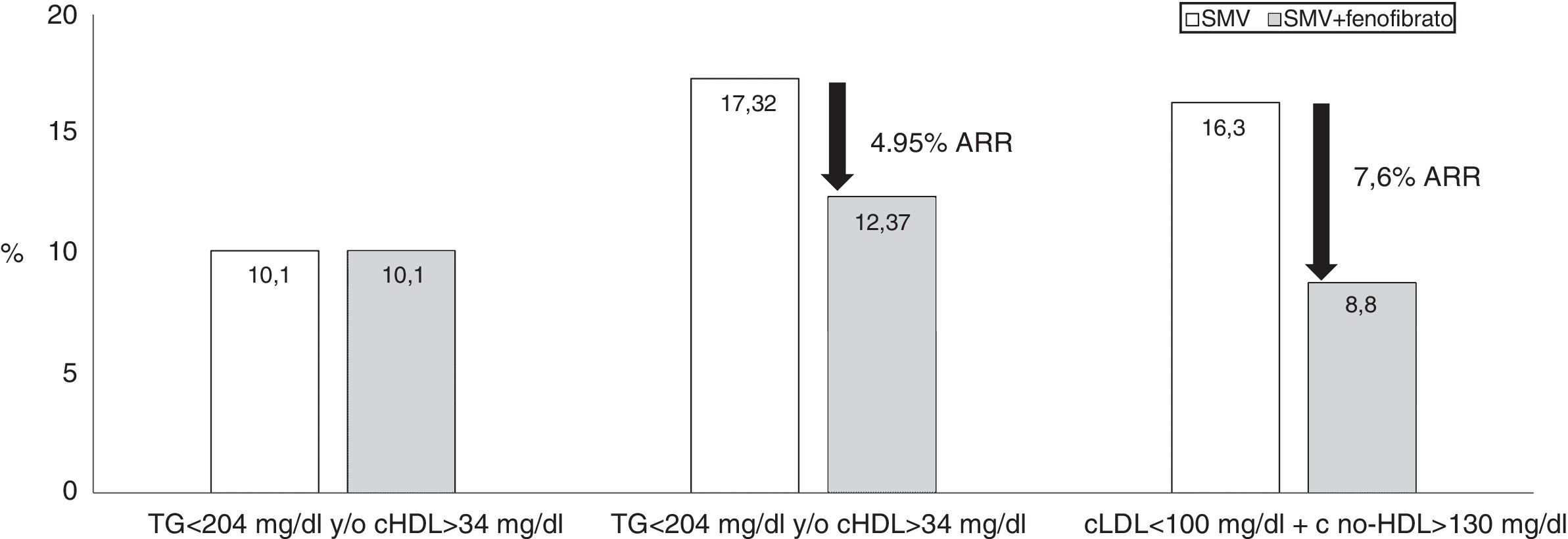
Para la reducción de TG, disponemos de un fármaco que ha demostrado cierta eficacia sobre el riesgo residual: el fenofibrato97. Por otra parte, cuando se asocia a estatinas, la seguridad de esta asociación ha quedado suficientemente probada en diferentes estudios clínicos98-100. Con esta asociación se comprueba, además de la reducción de las cifras de cLDL, un claro descenso de los niveles de TG y un incremento del cHDL. La combinación de simvastatina y fenofibrato en el estudio Action to Control Cardiovascular Risk in Diabetes (ACCORD) fue la primera descripción de la eficacia de esta asociación, si bien no redujo significativamente los episodios cardiovasculares no fatales y la muerte cardiovascular en pacientes diabéticos frente a la simvastatina en monoterapia. Sin embargo, los pacientes del grupo de DA presentaron un riesgo relativo un 70% más elevado de episodios cardiovasculares graves en comparación con la población diabética sin dislipidemia, a pesar de alcanzar un nivel medio de cLDL de 80mg/dl (fig. 3). Un análisis específico demostró que en pacientes con TG elevados (>200mg/dl) y cHDL disminuido (<34mg/dl) el RCV relativo se redujo en un 31% en el grupo de terapia combinada frente a la monoterapia con simvastatina101. Para prevenir un episodio cardiovascular es necesario tratar durante 5años tan solo a 20 pacientes con la combinación (la reducción del riesgo absoluto resultó 4,95%). El estudio Fenofibrate Intervention and Event Lowering in Diabetes Study fue diseñado para evaluar el efecto a largo plazo del fenofibrato sobre episodios cardiovasculares en pacientes con DM2102. Participaron 9.795 pacientes, con edad entre 50 y 75 años. No se produjeron reducciones significativas en los objetivos primarios de mortalidad, pero el tratamiento con fenofibrato disminuyó los episodios cardiovasculares totales. Los pacientes más beneficiados fueron los que presentaron TG>150mg/dl y un cHDL<40mg/dl en varones y 50mg/dl en mujeres. En este estudio se demostró además un efecto beneficioso del fenofibrato en el riesgo microvascular, con una reducción en la progresión de la oligoalbuminuria, en el desarrollo y progresión de la retinopatía y en el riesgo de amputación en extremidades inferiores. En consecuencia, para abordar la reducción del riesgo residual, especialmente en pacientes con DA en los que parece necesario contemplar la optimización de los resultados terapéuticos en la tríada lipídica (cLDL, cHDL y TG) eventualmente con una combinación de estatinas y fenofibrato, tal y como se ha comprobado hasta este momento103.
, pero con c-no-HDL > 130 mg/dl. Los eventos cardiovasculares mayores fueron: muerte cardiovascular, infarto de miocardio no fatal o accidente vascular cerebral. Fuente: Modificado de The ACCORD Study Group101.")
Estudio Accord-Lipid: 5.518 pacientes con DM2, seguimiento de 4,7 años. La presencia de dislipidemia aterogénica incrementó un 70% el riesgo de eventos cardiovasculares mayores. La asociación simvastatina y fenofibrato redujo significativamente el riesgo en sujetos con cLDL en rango (<100 mg/dl), pero con c-no-HDL > 130 mg/dl. Los eventos cardiovasculares mayores fueron: muerte cardiovascular, infarto de miocardio no fatal o accidente vascular cerebral. Fuente: Modificado de The ACCORD Study Group101.
Otro fármaco con capacidad de reducción de TG hasta un 25% e incremento de la concentración de cHDL entre un 15 y un 25%104 es el ácido nicotínico o niacina que, asociado a una estatina, comporta potenciales beneficios para la reducción del riesgo residual de origen lipídico105. La cumplimentación de este fármaco suele ser irregular debido a sus efectos secundarios, principalmente la rubefacción. El ácido nicotínico puede reducir la sensibilidad a la insulina, lo que favorece el desarrollo de DM2 en sujetos predispuestos o empeora el control metabólico en sujetos diabéticos106. En el estudio AIM-HIGH, realizado en pacientes con ECV aterosclerótica y niveles de cLDL de menos de 70mg/dl, no se encontró beneficio clínico a la asociación de niacina con estatinas durante un período de seguimiento de 36 meses, a pesar de mejoras en los niveles de cHDL y TG107. En el estudio HPS-THIVE, realizado en pacientes con enfermedad vascular aterosclerótica, la adición de niacina-laropiprant de liberación prolongada al tratamiento con estatinas no redujo significativamente el riesgo de eventos vasculares mayores, pero sí aumentó el riesgo de eventos adversos graves108. En otros estudios se ha comprobado una reducción de la mortalidad coronaria entre un 12 y un 14%109 con el uso de niacina y estatina, y se ha demostrado un descenso del grosor íntima-media carotídea superior al obtenido con la intensificación de estatina en monoterapia110. A pesar de los potenciales efectos beneficiosos demostrados en los estudios multicéntricos referidos, debido a sus efectos secundarios, el uso de este fármaco es prácticamente inexistente en la actualidad.
Por último, entre los fármacos indicados para el descenso de TG se encuentran los ácidos grasos omega-3 administrados a dosis de 3-4g/día, que reducen significativamente los TG y las LRTG, tanto en monoterapia como asociados a estatina111,112. En prevención secundaria, la utilización de los ácidos grasos omega-3ha demostrado una reducción de la morbimortalidad cardiovascular113: incluso con la dosis de 1g/día reduce la mortalidad y la hospitalización de causa cardiovascular en pacientes con insuficiencia cardíaca114. Sin embargo, diferentes metaanálisis posteriores han demostrado que el tratamiento con ácidos grasos omega-3 no se asocia con un efecto protector sobre los principales eventos cardiovasculares, mientras que ejerce efectos beneficiosos en la reducción de la muerte por causas cardíacas, muerte súbita cardíaca y muerte por todas las causas115,116.
Finalmente, la acción farmacológica para incrementar el cHDL está representada por los inhibidores de la PTEC. Entre los estudios realizados con estos fármacos, destacan los realizados con torcetrapid (ILLUSTRATE, RADIANCE, ILLUMINATE): los 3debieron ser suspendidos a los 18 meses por aumentar un 25% los eventos cardiovasculares y presentar un 40% más de muertes por dicha causa en la rama activa117. El estudio dal-OUTCOMES, realizado con dalcetrapid asociado a estatinas en pacientes con síndrome coronario agudo, también se interrumpió por falta de eficacia en la rama activa118. El estudio REVEAL, con anacetrapib, demostró beneficios cardiovasculares a partir del tercer año, sin reducción de la mortalidad119. El evacetrapib también consigue mejorar el perfil lipídico, pero no disminuye eventos cardiovasculares frente a placebo120.
En general, todas las acciones farmacológicas destinadas a reducir el RCV residual de origen lipídico, teniendo como dianas los TG o el cHDL, han tenido resultados escasamente beneficiosos o incluso contradictorios, excepto en determinados análisis post hoc, en poblaciones concretas. Por esta razón, ninguna guía actual reconoce como objetivos terapéuticos los TG y el cHDL.
ConclusiónDisponemos de una ingente evidencia científica de la influencia del cLDL en el RCV, así como de su relación en la morbimortalidad cardiovascular. Sin embargo, el concepto de riesgo residual de origen lipídico comprende al persistente y elevado RCV, una vez conseguido el objetivo del cLDL en los valores aceptados por la mayoría de las sociedades científicas. Los factores responsables de este riesgo residual lipídico comprenden los TG y las lipoproteínas que los transportan (LRTG), además del descenso de cHDL y el incremento de las partículas LDL. Estas alteraciones lipídicas están definidas con el concepto de DA. Para su identificación y plan de intervención nos valemos de sencillos cálculos, tales como el c-no-HDL o el colesterol remanente, que han sido incorporados como objetivos terapéuticos en la mayoría de las guías clínicas, especialmente el primero. Por último, además de modificaciones en la dieta y del aumento de ejercicio físico, tenemos un escaso arsenal terapéutico para combatir dicho riesgo: solo disponemos del fenofibrato como fármaco eficiente en pacientes con DA. Los nuevos fármacos no estatinas relacionados con el incremento de receptores LDL parecen tener un efecto eficaz, pero la limitación de su uso en la clínica habitual reduce el potencial beneficio en la población con riesgo residual lipídico elevado.
Conflicto de interesesLos autores confirman la ausencia de conflicto de intereses.