




Acute myocardial infarction (AMI) is one of the major causes of mortality and morbidity worldwide. Despite the efforts being made, there is a lack of early markers for the prevention, diagnosis and treatment of ischemic syndromes. Proteomic expression profiling technologies are a highly important tool for research into new serum biomarkers for the diagnosis and prognosis of acute coronary syndromes.
MethodsSerum samples were sub-fractionated with different methods for the depletion of high-abundance proteins. The low-abundance fraction was analyzed by two-dimensional electrophoresis (2-DE), followed by protein identification with mass-spectrometry (MALDI-TOF). The proteomic profiles of serum samples from AMI patients and controls were analyzed and compared.
ResultsThrough depletion of six high-abundance proteins in 2-DE analysis of serum samples, 569 spots were detected, of which 131 spots were only detected in the AMI group and 27 were only detected in controls. The comparative analysis between AMI-patients and controls revealed a group of differential protein spots involved in seven different biological functions. The main changes were found in proteins involved in the immune system and lipid metabolism.
ConclusionsIn this study, by using a 2-DE differential approach, we developed a highly reproducible methodology for the analysis of coordinated changes in serum proteome patterns that occur within the first 6hours after the onset of an AMI.
El infarto agudo de miocardio (IAM) es una de las mayores causas de mortalidad y morbilidad en el mundo. A pesar de todos los esfuerzos hay una falta de marcadores para la prevención, el diagnóstico y el tratamiento de la fase temprana de los síndromes isquémicos. Las técnicas proteómicas son una herramienta muy importante para la búsqueda de nuevos biomarcadores para el diagnóstico y el pronóstico de los síndromes coronarios agudos.
MétodosLas muestras de suero se sub-fraccionaron mediante diferentes métodos para eliminar las proteínas mayoritarias. La fracción de proteínas de menor abundancia se analizó mediante electroforesis bidimensional (2-DE), seguida de la identificación de proteínas mediante espectrometría de masas (MALDI-TOF). Se analizó y comparó el patrón proteómico de los pacientes IAM y el grupo control.
ResultadosMediante la eliminación de las seis proteínas mayoritarias en el análisis por 2-DE se detectaron un total de 569 spots, de los cuales 131 sólo se detectaron en el grupo infarto y 27 sólo en el grupo control. El análisis comparativo entre los pacientes IAM y los controles reveló un grupo de spots proteicos con un patrón de distribución diferencial entre ambos grupos. Estos spots diferenciales pertenecen a siete grupos diferentes en función de su papel biológico, de los cuales los cambios más importantes se ven en las proteínas involucradas en el sistema inmune y en el metabolismo lipídico.
ConclusionesEn el presente estudio, mediante el uso de técnicas de 2-DE hemos desarrollado una metodología altamente reproducible para el análisis de los cambios coordinados que se dan durante las seis primeras horas desde el inicio de un evento.
Ischemic atherothrombotic syndromes induce structural and functional modifications that are reflected in serum levels of proteins and other biomarkers. However, biomarkers characterization is nowadays incomplete and their prognostic value is not clear. Increased concentrations of inflammatory biomarkers such as C reactive protein (CRP), serum amyloid A, myeloperoxidase and interleukin-6 (IL-6) are detectable in a substantial proportion of patients with acute coronary syndromes. Several biomarkers of myocardial ischemia are under investigation, ischemia-modified albumin (IMA) is among the most thoroughly studied of these markers.1 There are also biomarkers for the detection of cardiac injury, from those ones the protein of choice is troponin.2
Some of those biomarkers lack of consensus because, up to now, it is not clear whether their measurement would be useful in the diagnosis and prognosis of ischemic atherothrombotic syndromes.
Despite the success of cardiac troponins as markers of myocardial injury there is still a need for the development of early markers for prevention, diagnosis and treatment of ischemic syndromes.
Proteomics is one of the most important strategies for the study of complex protein associations. By using proteomic techniques we can determine modifications in protein structure, expression levels, and post-translational modifications that may be associated to the presence of disease. Indeed, proteins are excellent targets for disease diagnostic and prognostic markers and to develop therapeutic strategies. Proteomic techniques are a good tool for the analysis of the protein content in a determined sample.3
Serum has the advantage of being one of the most accessible sources for biomarker discovery, but also has the disadvantage of its great complexity as it has a lot of different proteins in a very dynamic range of concentrations. Near 85% of serum proteome is represented by a reduced group of proteins that are known as the high abundant fraction and usually have no interest in proteomic analysis as they are very well characterized. Potential serum biomarkers are in very low abundance and so they are masked by high abundant proteins such as albumin and IgGs.
The aim of this study is to characterize the serum proteomic profile in healthy individuals and acute myocardial infarction patients by using two different subfractionation methods.
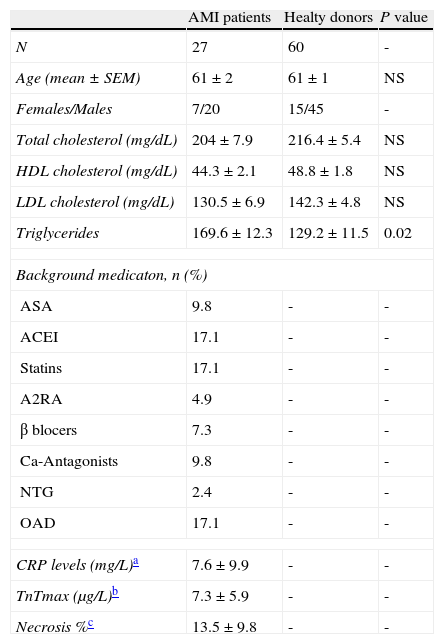
Materials and methodsStudy populationThe study population comprised 27 new-onset AMI patient (20 men and 7 women; mean age: 61±2 years) who were admitted with chest pain and suspected of ACS at the Emergency Room of Santa Creu i Sant Pau Hospital.
At the emergency department, routine diagnostic procedures were applied to establish the onset of symptoms as accurately as possible (i.e. description of chest pain, pulmonary edema, severe dyspnea, and syncope). In addition to the general patient history, clinical examination, 12-lead ECG, and laboratory tests were also run to characterize AMI-patients. All AMI-patients showed (1) typical chest pain lasting more than 30minutes; (2) ST segment elevation >0.2mV in at least 2 contiguous leads; (3) admission to the hospital within the first 6hours after chest pain onset; (4) normal serum CK and CKMB levels at admission; (5) negative troponin T at admission (excluding subacute myocardial infarction); (6) sinus rhythm. Exclusion criteria were a previous documented or suspected myocardial infarction and antithrombotic treatment because of the AMI onset before arriving to the emergency room and time of blood collection. Delayed contrast-enhanced (CE) cardiovascular magnetic resonance studies were performed within the first week after acute ST-segment elevation AMI in all patients to evaluate the MI location and the necrotic lesion. The control group included sixty healthy individuals (45 men and 15 women; mean age: 61±1 years) who attended to a routine health check. Background description of AMI-patients and control individuals are listed in Table 1.
Background description of AMI-patients and healthy donors.
| AMI patients | Healty donors | P value | |
| N | 27 | 60 | - |
| Age (mean±SEM) | 61±2 | 61±1 | NS |
| Females/Males | 7/20 | 15/45 | - |
| Total cholesterol (mg/dL) | 204±7.9 | 216.4±5.4 | NS |
| HDL cholesterol (mg/dL) | 44.3±2.1 | 48.8±1.8 | NS |
| LDL cholesterol (mg/dL) | 130.5±6.9 | 142.3±4.8 | NS |
| Triglycerides | 169.6±12.3 | 129.2±11.5 | 0.02 |
| Background medicaton, n (%) | |||
| ASA | 9.8 | - | - |
| ACEI | 17.1 | - | - |
| Statins | 17.1 | - | - |
| A2RA | 4.9 | - | - |
| β blocers | 7.3 | - | - |
| Ca-Antagonists | 9.8 | - | - |
| NTG | 2.4 | - | - |
| OAD | 17.1 | - | - |
| CRP levels (mg/L)a | 7.6±9.9 | - | - |
| TnTmax (μg/L)b | 7.3±5.9 | - | - |
| Necrosis %c | 13.5±9.8 | - | - |
ASA: acetylsalicylic acid; ACEI: angiotensin-converting enzyme inhibitors; A2RA: angiotensin 2 receptor antagonists; NTG: nitroglycerine; OAD: oral antidiabetic drugs.
The Ethics Committee of the Santa Creu i Sant Pau Hospital approved the project and the studies were conducted according to the principles of Helsinki's Declaration. All participants gave written informed consent to take part in the study.
Blood collection and sample preparation- •
Blood samples obtention. Venous blood samples of AMI-patients, before starting any medication, and control individuals were collected to prepare serum that was aliquoted and stored at −80°C.
- •
Serum subfractionation. For proteomic studies, serum samples were sonicated (six cycles of 15seconds each) in ice4 and filtrated (0.22μm) by centrifugation to avoid the presence of impurities. Highly abundant proteins were depleted using two different depletion methods one that uses an antibody affinity resin to selectively remove albumin and IgGs (Albumin and IgG Removal Kit from GE Healthcare) and another that removes the six most abundant serum proteins using a specific affinity cartridge with binding capacity for albumin, IgGs, IgAs, transferrin, α1 antytripsin and haptoglobin (Multiple Affinity Removal Spin Cartridge, Agilent Technologies) as reported by the providers. Serum depleted samples (called the total serum fraction) were concentrated and desalted by centrifugation with 5 kDa cut-off filter devices and sample buffer was exchanged to a urea containing buffer (8M Urea, 2% Chaps). Protein concentration in the serum extracts was measured with 2D-Quant Kit (GE Healthcare). All processed samples were stored at –80°C until used.
C-reactive protein (CRP), lipids (Total-, HDL-, LDL-cholesterol, and triglycerides), kidney parameters (urea and creatinine), liver parameters (bilirubin, GOT and GPT) and T-troponin levels were measured by standard laboratory methods.
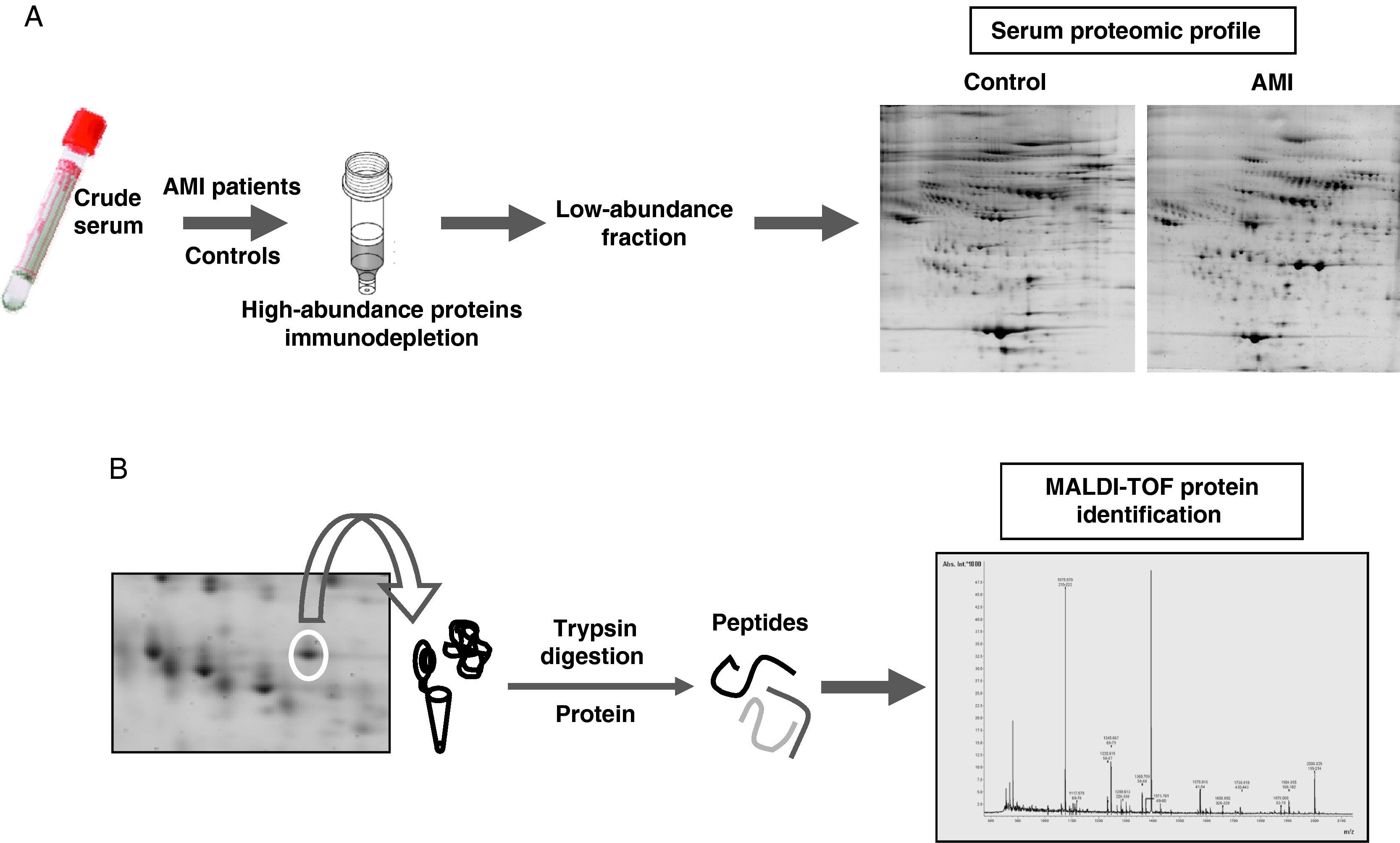
Proteomic analysisThe proteomic analysis of serum samples was performed by two-dimensional electrophoresis (Figure 1A) followed by mass spectrometry identification (Figure 1B).
- •
Two-dimensional gel electrophoresis (2-DE). For analytical and preparative gels, respectively, a protein load of 120μg and 300μg protein were dissolved in the rehydration buffer (8M urea, 2% chaps, 0.2% ampholytes, 1.6% DTT and 0.002% bromophenol blue) soluble extracts was applied by cup-loading to 18-cm dry strips (pH 4-7 linear range, GE Healthcare). Isoelectrofocusing was performed in a Protean IEF Cell (BioRad) at the following conditions: (1) linear step at 250V for 45min; (2) linear step at 500V for 1h; (3) linear step at 1000V for 1h; (4) linear step at 4000V for 1h; (5) linear step at 10,000V for 1h; (6) linear step at 10,000V until reaching 70,000V. After the first dimension strips were equilibrated 15min in an equilibration buffer (6M urea, 2% SDS, 50mM Tris-HCL pH 8.8 and 30% glycerol) containing 1% DTT and 15min in the same buffer with 2.5% iodoacetamide.
The second dimension was resolved in 10% SDS-PAGE in an Ettan Dalt six electrophoresis unit (GE Healthcare) at 5 w/gel during the first 30minutes and afterwards at 17 w/gel. Gels were developed by fluorescent staining (analytical gels) or comassie blue (preparative gels). For each independent experiment, 2-DE for protein extracts from controls and patients were processed in parallel to guarantee a maximum of comparability. Each 2-DE run was at least repeated twice. Analysis for differences in protein patterns was performed with the PD-Quest 8.0 (BioRad), using a single master that included all gels of each independent experiment. Each spot was assigned a relative value that corresponded to the single spot volume compared to the volume of all spots in the gel, following background extraction and normalization between gels.
- •
Mass spectrometry analysis. Protein spots of interest were excised from 2-DE gels, washed (25mM Ambic), dehydrated (25mM Ambic/50% ACN followed by 100% ACN), dried, and enzymatically digested with one gel volume of sequence-grade modified porcine trypsin (Promega). Peptides from in-gel-trypsin digestion were desalted and concentrated by ZipTipU-C18 (Millipore), mixed 1:1 with 5mg/mL α-cyano-4-hydroxy-cinnamic, and spotted on a stainless steel mass spectrometry slide. Protein identification was performed by peptide-mass fingerprinting using an Ettan MALDI-TOF Pro (matrix-assisted laser desorption/ionisation time-of-flight mass spectrometer, GE-Healthcare) operating in delayed extraction/reflector mode. MALDI-generated mass spectra were internally calibrated using trypsin autolysis products, Ang III (angiotensin III), and ACTH (adrenocorticotropic hormone) peaks. The peptide masses were searched against the National Center for Biotechnology Information nonredundant mammalian database using ProFound™ and confirmed using a Mascot 2.3 search from Matrixscience, selecting the SwissProt database. For the present study, protein identification was based on the measurement with a Mascot score higher than 55 and a minimum coverage of 20%. Minimal expectation for valid identification was <0.005 and P<0.05.
 Experimetal workflow of serum sample sub-fractionation and the analysis by 2-DE of the low abundance fraction in acute myocardial infarction patients and the control group. (B) Working scheme of 2-DE spots excision, trypsin digestion and peptide mass fingerprint analysis by mass spectrometry (MALDI-TOF) for protein identification.")
(A) Experimetal workflow of serum sample sub-fractionation and the analysis by 2-DE of the low abundance fraction in acute myocardial infarction patients and the control group. (B) Working scheme of 2-DE spots excision, trypsin digestion and peptide mass fingerprint analysis by mass spectrometry (MALDI-TOF) for protein identification.
Data are expressed as mean and standard error of the mean ± SEM unless stated. N indicates the number of subjects tested. Statistical analysis for differences between control and AMI groups were performed by the ANOVA single factor or including covariates, Fisher PSLD as post-hoc analysis, and the Student's t-test or the non-parametric Mann-Whitney test, as indicated. Correlations between variables were determined single and multiple regression models. A P value ≤0.05 was considered significant.
ResultsClinical characteristics of the study populationThere was no significant difference in age, sex and cholesterol levels (Total-, HDL- and LDL-cholesterol) between AMI-patients and controls. AMI-patients showed significant higher levels of triglycerides (P=0.02), urea (P=0.03), creatinine (P<0.001), and also significant higher levels of hepatic parameters as bilirubin (P<0.001), GOT (P<0.001), and GPT (P<0.001) when compared to controls. No T-troponin levels were detected at arrival to the hospital.
The high abundance fraction interferes in the detection of serum proteomeAs shown in Figure 2 the same serum samples were analyzed using two different depletion methods in parallel and compared to crude serum.
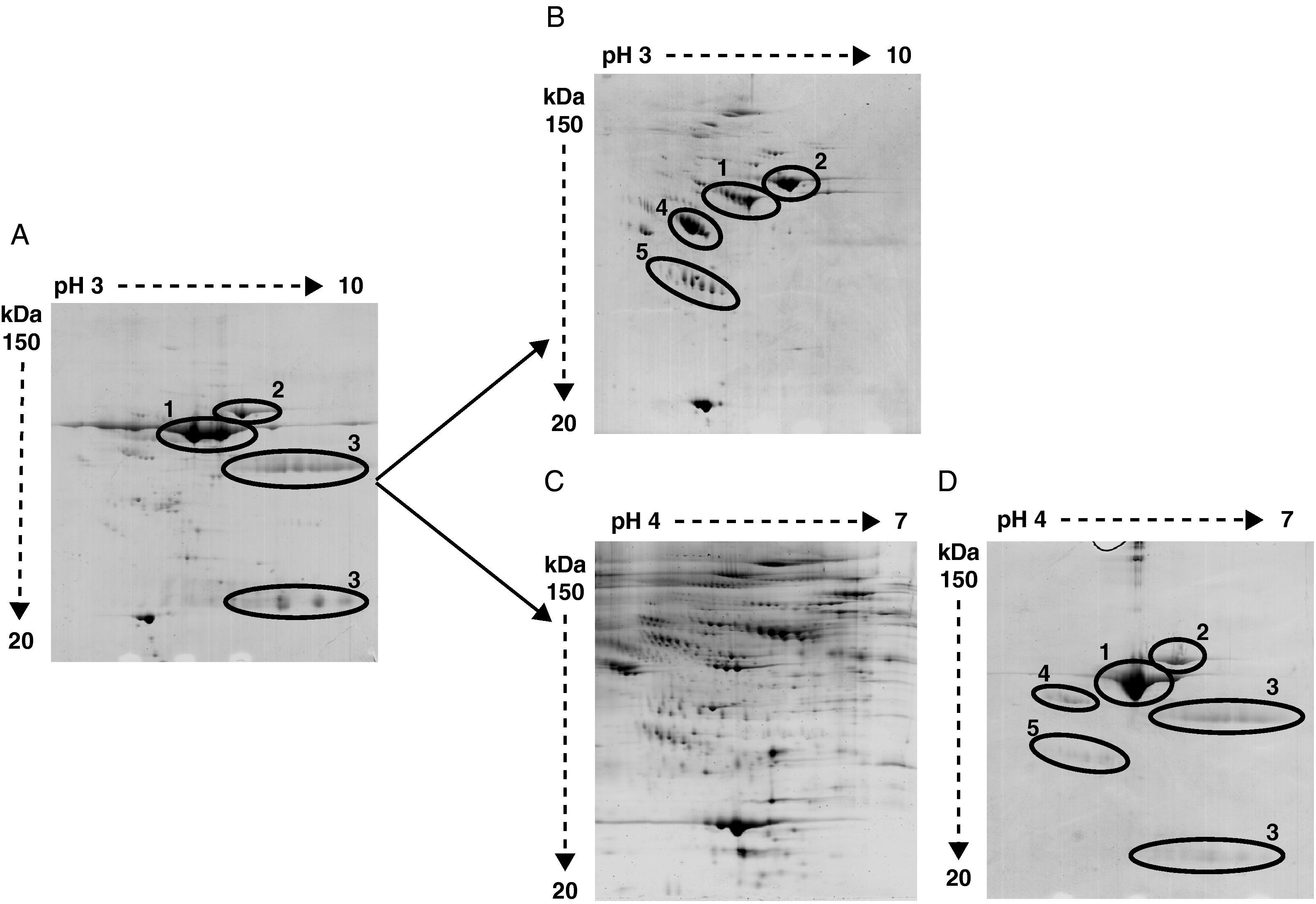
 crude serum, (B) serum depleted of albumin and IgGs, (C) serum depleted of six high-abundant proteins (albumin, IgGs, IgAs, transferrin, haptoglobin and α1-antitrypsin), and (D) the retained fraction containing those six high-abundant proteins. In crude serum only albumin (1), transferrin (2) and IgGs heavy and light chain (3) are detected. After albumin and IgGs depletion α1-antitrypsin (4) and haptoglobin (5) are apparent. After six high abundant protein depletion the amount of detected proteins is increased. The retained fraction containing the six high-abundant proteins show a very similar profile of that obtained with crude serum.")
Representative image of 2-DE of (A) crude serum, (B) serum depleted of albumin and IgGs, (C) serum depleted of six high-abundant proteins (albumin, IgGs, IgAs, transferrin, haptoglobin and α1-antitrypsin), and (D) the retained fraction containing those six high-abundant proteins. In crude serum only albumin (1), transferrin (2) and IgGs heavy and light chain (3) are detected. After albumin and IgGs depletion α1-antitrypsin (4) and haptoglobin (5) are apparent. After six high abundant protein depletion the amount of detected proteins is increased. The retained fraction containing the six high-abundant proteins show a very similar profile of that obtained with crude serum.
Bidimensional electrophoresis of crude serum only displayed two big spots corresponding to albumin and transferrin and two areas of blurred spots identified as heavy and light chains of IgGs respectively (Figure 2A).
After removing albumin and IgGs other proteins like transferrin, α1-antytripsin and haptoglobin turned out to be the most detected proteins and prevent other less abundant proteins from being detected (Figure 2B). The identification by MALDI-TOF analysis of protein spots obtained after removing albumin and IgGs revealed 25 non-redundant proteins (data not shown). The immunodepletion of albumin, IgGs, IgAs, transferrin, haptoglobin and α1 antytripsin allowed the further identification of 90 non-redundant low abundance proteins not detected with the first method (Figure 2C). A total of 569 spots were detected in serum samples. In the AMI group a total of 542 spots were detected. The control group depicted 438 total spots, from which 411 spots were also detected in the AMI group. There were 131 spots only present in the AMI group and 27 spots only detected in the control group. The detected serum spots were mainly in the pI range between 4 and 7, and in a molecular weight range between 150 and 20 kDa. The identified proteins are involved in different processes listed in Table 2. The proteomic profile of the retained fraction containing those six high-abundant proteins was very similar to the crude serum profile (compare Figure 2A with 2D). The identification by mass spectrometry of the detected spots in the retained fraction only revealed the presence of high-abundant proteins.
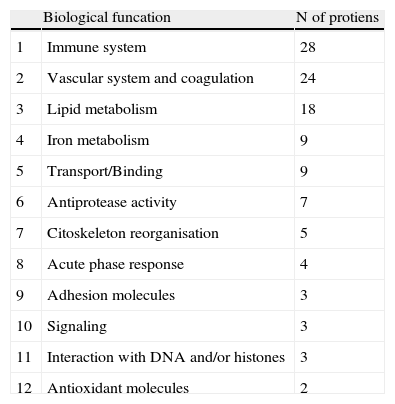
Groups of proteins identified in serum proteome.
| Biological funcation | N of protiens | |
| 1 | Immune system | 28 |
| 2 | Vascular system and coagulation | 24 |
| 3 | Lipid metabolism | 18 |
| 4 | Iron metabolism | 9 |
| 5 | Transport/Binding | 9 |
| 6 | Antiprotease activity | 7 |
| 7 | Citoskeleton reorganisation | 5 |
| 8 | Acute phase response | 4 |
| 9 | Adhesion molecules | 3 |
| 10 | Signaling | 3 |
| 11 | Interaction with DNA and/or histones | 3 |
| 12 | Antioxidant molecules | 2 |
Serum samples (after removal of the six most abundant proteins) from 27 AMI-patients were analyzed individually and samples of healthy donors were analyzed in 6 pools of 10 individuals each in order to obtain a more representative pattern of a healthy population. All samples were run in duplicates by 2-DE and proteins were identified by MALDI-TOF mass spectrometry.
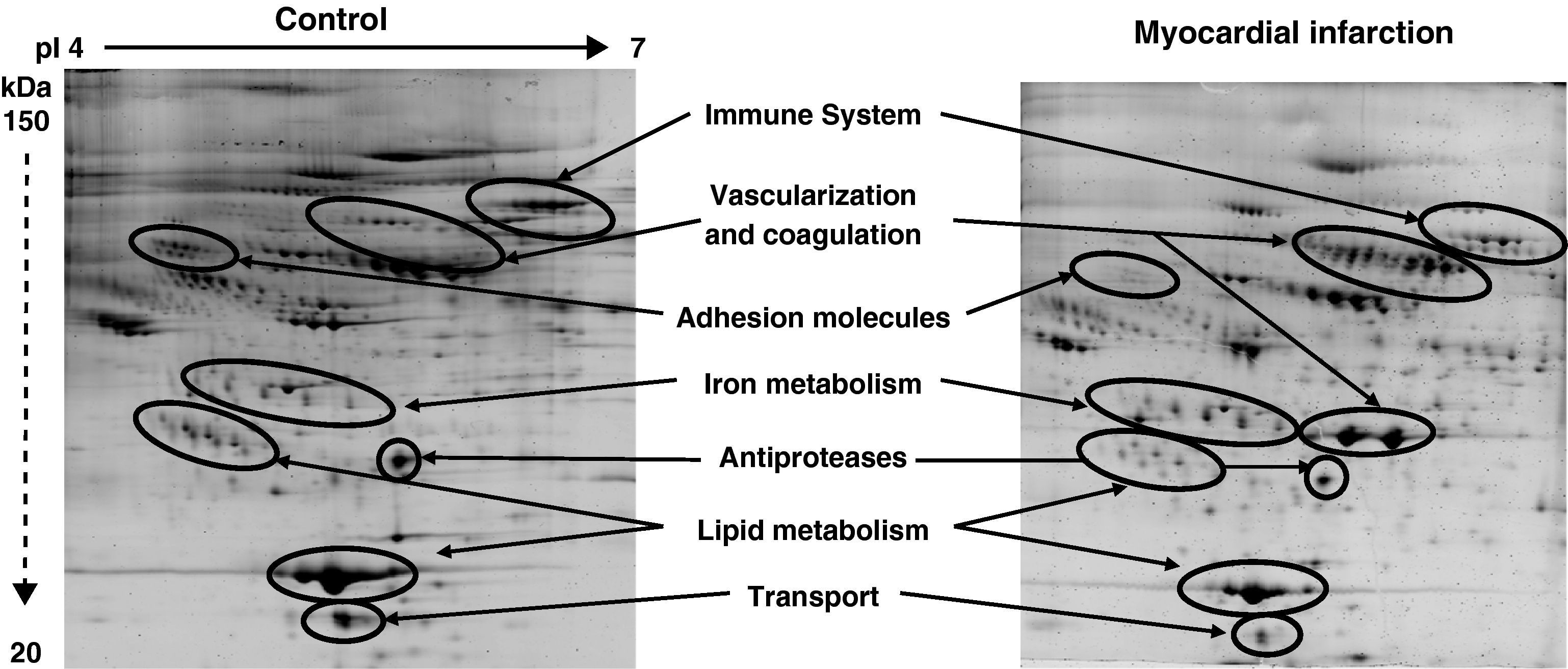
Among the 569 detected serum spots, 270 showed a statistically significant differential expression profile between AMI patients and control individuals, being 127 of them identified as 28 non-redundant proteins involved in seven different biological functions (Figure 3).

Representative 2-DE image of control and myocardial infarction serum samples. There are several spot proteins with differential expression profile between AMI patient and control individuals. These differential proteins are involved in seven different biological functions: immune system, vascularisation and coagulation, adhesion molecules, iron metabolism, antiproteases, lipid metabolism, and transport proteins.
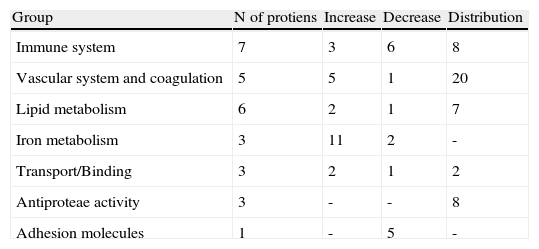
From the identified spots with a differential expression profile, 23 showed at least a two-fold increase in their intensity, 16 depicted at least a two-fold decrease, and 45 showed changes in their distribution profile in AMI patients because changes in molecular weight and/or pI, presumably due to post-translational modifications (Table 3).
Groups of proteins with differential profile in AMI patients compared to healthy donors.
| Group | N of protiens | Increase | Decrease | Distribution |
| Immune system | 7 | 3 | 6 | 8 |
| Vascular system and coagulation | 5 | 5 | 1 | 20 |
| Lipid metabolism | 6 | 2 | 1 | 7 |
| Iron metabolism | 3 | 11 | 2 | - |
| Transport/Binding | 3 | 2 | 1 | 2 |
| Antiproteae activity | 3 | - | - | 8 |
| Adhesion molecules | 1 | - | 5 | - |
There is an increasing need for the identification of new biomarkers in order to obtain tools for an early detection and prognosis of atherothrombotic events such as acute myocardial infarction. To this aim serum proteomics reveals as a leading technology for the research and characterization of new biomarkers. Accordingly, in this study we have used proteomic analysis to identify new changes in serum proteome in association to the clinical manifestation of cardiovascular disease such as AMI.
Serum is a very rich source for biomarker discovery but because of its great complexity it must be sub-fractionated prior to proteomic analysis. Therefore to analyze serum samples by bidimensional electrophoresis a previous step to remove the high abundance proteins is essential. Years ago the depletion of human albumin was performed via Cibacron Blue columns.5 In case of IgGs, resins of immobilized protein A or G have been widely used.6 But these methods lacked of specificity and reproducibility. Nowadays there are several methods based on columns containing a mixture of antibodies for the depletion of various high abundance proteins at the same time.
In this work the selected depletion method was the one removing six high abundance proteins (albumin, transferrin, IgGs, IgAs, haptoglobin and α1 antitrypsin). By using this immunodepletion method we have developed a highly reproducible technique for the identification and analysis of a wide range of low abundance proteins from serum proteome. The use of these antibody-based columns has reduced the nonspecific binding during depletion, in fact by 2-DE analysis we did not detect nonspecific proteins in the retained fraction. Unfortunately, we can not exclude that some low-abundance proteins, not detectable by 2-DE analysis were lost during the depletion protocol. Nevertheless, our methodological approach represents an improvement in protein detection compared to previous studies.7
Early systemic changes after the onset of an AMI have not been fully characterized and therefore there is a dearth of biomarkers that could indicate early ischemic damage. The analytical power of modern proteomic technologies could help to identify these systemic changes and therefore facilitate our understanding of the pathophysiology of cardiovascular diseases.8 In the present study, by applying proteomic technologies, we have detected significant changes in groups of proteins involved in several different biological functions such as: immune system, vascular system and coagulation, lipid metabolism, iron metabolism, transport/binding, antiprotease activity, and adhesion molecules.
Among the proteins showing a differential profile between AMI-patients and control individuals there are seven immune system related proteins. There are several evidences of the relevance of immune response processes and related molecules in atherothrombosis.9,10
In this study we have found that acute myocardial infarction is related to a differential proteomic profile in proteins involved in lipid metabolism. Some authors11 suggest, for example, that the relevance of HDL in cardiovascular diseases is not through its levels (HDL-cholesterol levels) in plasma but through its composition, structure and function. Therefore, the more in depth study of proteins involved in lipid metabolism such as lipoproteins may lead to a better understanding of their role in atherothrombotic diseases.
In AMI-patients differential proteins did not only showed increased or decreased intensity values in comparison to healthy donors, but also depicted changes in their distribution pattern presumably due to post-translational modifications or the presence of different protein isoforms. These protein modifications, such as phosphorylations and glycosylations, play an important role in the molecular pathology of a disease.12 The specific study of serum phospho13 and glycoproteomes14 could represent and important source for biomarker discovery. The most important post-translational modification in serum proteome is glycosylation. In fact there are several studies relating differential glycosylation states of serum proteins in relation to specific diseases.15–17
In this study, by using a 2-DE differential approach we have developed a highly reproducible methodology for the analysis of coordinated changes of serum proteome that exist within the first six hours after the onset of an acute myocardial infarction. Further studies are ongoing in our group in order to determine the specific changes of the proteins involved in those biological functions that have shown to be differentially represented in the proteomic profile of patients in the early phase after an AMI.
DisclosuresNo conflicts to disclose.
This work has been possible due to the funds provided by SAF 2006/10091 to L.B., CIBER OBENU CB06/03 to L.B., FIS-PI071070 to T.P., REDINSCOR RD06/0003/00 to J.C. and REDINSCOR RD06/0003/0015 to T.P. and TERCEL to L.B. from Instituto Carlos III; “Fundación Lilly” and “Fundación Jesus Serra”.
This work, presented as an oral communication entitled, “Apolipoproteína J en pacientes con infarto agudo de miocardio de nueva presentación” during the XXIII Congreso Nacional de la SEA in Córdoba 2010, has received the award “Mención Especial” (Ref. ME2010-6.30).