





So far, most cases of hypercholesterolaemia (60-80%) are attributed to pathogenic variants in the LDLR gene. Only 1-5% of cases are caused by variants in the APOB gene, and 0-3% by variants in the PCSK9 gene. There is a large variety in known pathogenic mutations of the LDLR gene, while for those affecting the APOB gene, the highest incidence is p.Arg3527Gln, described predominantly in Central European and North American populations. In the Iberian Peninsula the predominant gene affected is that of the LDL receptor, similar to the rest of the world, with the involvement of the APOB gene being described in individuals from the northwest, and anecdotal in the rest of the territory. A genetics analysis was performed on the population attending the first year of a lipid clinic in southwestern Spain with a 6-point score from the Dutch lipid clinics. The genetic, biochemical and clinical findings are described. The first findings show indications of a possible higher prevalence of patients with mutation in the APOB gene compared to other territories. Historical evidence is presented that could give a possible explanation to this, thus supporting the assumption.
Hasta el momento, la mayor parte de los casos de hipercolesterolemia familiar (60-80%) se atribuyen a variantes patogénicas en el gen LDLR. Sólo un 1-5% de los casos se produce por variantes en el gen APOB y un 0-3% por variantes en el gen PCSK9. Existen gran variedad en mutaciones patogénicas conocidas del gen LDLR mientras que, de las que afectan al gen APOB, la de mayor incidencia es p.Arg3527Gln, descrita predominantemente en poblaciones de Centroeuropa y América del Norte. En la península Ibérica el gen predominante afectado es el del receptor de LDL, similar al resto del mundo, siendo la afectación del gen APOB descrita en individuos del noroeste y anecdótica en el resto del territorio. Analizamos genéticamente la población asistida en el primer año de una consulta de lípidos del suroeste de España con puntuación ≥6 puntos de las clínicas de lípidos holandesas y describimos los hallazgos genéticos, bioquímicos y clínicos. Los primeros hallazgos muestran indicios de una posible mayor prevalencia de pacientes con mutación en el gen APOB respecto a otros territorios. Encontramos hechos históricos que darían una posible explicación a este hecho, apoyando así dicha presunción.
Familial hypercholesterolaemia (FH) is a common genetic disorder characterised by increased circulating low-density lipoprotein cholesterol (LDLc), tendon xanthomas and premature heart disease. This is the most common genetic disease, with an estimated prevalence of approximately 1:250 according to the latest reviews.1 There is a dominant autosomal heredity pattern and they occur due to the presence of pathogenic variants in genes involved in the metabolism of the LDL receptor (LDLR).2 In most studies, the frequency of mutations detectable in patients with clinically defined or probable heterozygote FH (score in the Dutch lipid clinical levels network ≥6 points) is only 60%-80%, which indicates that a considerable proportion of patients with FH have a polygenic cause or the disease is caused by as yet unidentified genes.2 In 95% of the cases in which a genetic pathogenic change was identified, this was located in the LDLR gene. In the other positive genetic studies we found that 4%–5% of them had mutations in APOB and 1% had mutations in the gene which codified the proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein which induces degradation of the LDLR.
Later it was suggested that another 2 additional minority genes were causes of FH. On the one hand, in 2014 mutations which affected the gene STAP1 were described, which coded the signal transducing adaptor family member 1.3 However, recently Lamiquiz-Moneo et al.4 questioned this, demonstrating that there did not appear to be an aetiological role because there was an absence of correlation between genotype and phenotype. However, Cenarro et al.5 described the mutation p.(Leu167del) in the gene APOE as the cause of autosomal dominance in 3.1% of the subjects who had not presented mutation in other already known genes, such as LDLR, APOB or PCSK9.
FH presents with genetic heterogeneity since a very high number of causal variants have been described:>2000 in LDLR; 32 in APOB, 23 in PCSK9 and 1 in APOE.3
The most common mutation found in the APOB gene is a replacement of glutamine by arginine in position 3527 (p.Arg3527Gln), which shows impairment of the capacity for attachment to LDLR and reduction of LDL uptake.
The mutation p.Arg3527Gln was almost exclusively detected in Caucasian individuals (frequency in populations of European ascendency 1:500 to 1:700 individuals).6 Patients initially studied in Eastern Europe and North America, showed the same haplotypes at the ApoB locus. Their origin would come from common ancestors who lived at that time, in the Mesolithic period (10,000 to 6,000 years ago). This coincidence of haplotypes is repeated in the majority of subjects with the same mutation worldwide, with origins in central Europe and with the Alps and Pyrenees as geographical barriers for migration, presenting a low incidence in Mediterranean territories. Furthermore, this mutation was already present in the ancestors of the prehistoric period of the Mesolithic period in Celtic villages (Helvetii) which dominated the territory from the Rhine to the Main and who emigrated to the north of what is Switzerland today over 2000 years ago. 6,7
With regards to the known data in the Iberian Peninsula, it has been estimated that the prevalence of hypercholesterolaemic subjects with a familial defect of apolipoprotein B in the general Spanish population is very low (2.8×10−5). Most cases (84%) are located in a region of Celtic ascendency in the Northeast of Spain.6
The spectrum of mutations corresponding to APOB involvement has increased in recent years due, among other aspects, to next generation sequencing techniques.8 Recently, over 300 variants associated with FH have been studied but only 6% for them correspond to APOB. It is equally known that the mutations in the APOB gene do not have a 100% penetration and that the phenotype of patients is usually lighter than in patients with FH due to the LDLR mutations.
Most heterozygotes with APOB gene involvement respond well to medication which reduces the plasmatic levels of LDL on induction of receptor activity. It is also known that heart disease is rarely associated under 50 years of age, unlike cases with other vagrants which affect LDLR.6
We analysed the genetic particularities of the population covered by the lipids unit of the southeast of the Iberian Peninsula and their relationship with the regional historical particularities, together with their clinical and analytical findings.
Material and methodsPatientsA Spanish national Arian Project for FH screening was established in accordance with laboratory parameters (LDL≥250mg/dl), which our centre participated in. Patients were analysed from March 2018 until March 2019.
Firstly, those patients with secondary dyslipidaemia causes were ruled out, including hypothyroidism, nephrotic syndrome, advanced renal disease, hepatic cholestasis, excessive alcohol consumption, pregnancy and drugs (antiretroviral, anabolising, oestrogens, progestogens, glucocorticoids, tacrolimus and cyclosporine).A note was made for re-assessment to take place if LDLc levels continued to be high following resolution of the secondary causes. The patients who presented with diabetes were assessed in the practice since they presented with a very high cardiovascular risk, together with the possibilities of a diagnosis of FH coexisting.
The other patients were assessed in the practice, where a complete medical history was made with standard cardiovascular risk factors being collected (high blood pressure, diabetes, tobacco habit, etc.) and clinical and subclinical screening of cardiovascular disease. The Dutch Lipid Clinic Network9 criteria were applied to those with a score ≥6 points, and therefore with probable or certain diagnosis for HF. The genetic study was carried out, after obtaining their informed consent.
Geographic origin of patients corresponded to our hospital centre, situated in Southeast Spain, with subjects from different families who were unrelated.
Total blood samples were extracted for DNA studies and serum for the determination of biochemical parameters.
DNA extractionDNA extraction was undertaken from 5ml of peripheral anticoagulating blood with EDTA, using a commercial kit (Puregene: DNA isolation kit, Gentra Systems, MN.U.S.A.).
Next generation sequencing panelThe Lipid inCode® (Novartis®) panel was carried out in the authorised Lipid in Code laboratory (La Coruña, Spain). This panel is able to analyse the DNA promoter, sequence of the intron/exon junctions of the coding regions (±25pb) of the genes LDLR (NM_000527.4), APOB (NM_000384.2), PCSK9 (NM_174936.3), APOE (NM_000041.3), STAP1 (NM_012108.3;), LDLRAP1 (NM_015627.2) and LIPA (NM_000235.3).
The fortification of the sample into regions of interest was carried out with the Aiglent SureSelect QXT by Agilent (fortification solution designed and validated by Gencardio/GenIncode). The resulting libraries were sequenced massively in the MiSeq System ilumina platform, using chemistry based on clonal amplification and sequencing by synthesis technique. The genetic regions with a call rate <100% to a 30x coverage and associated variants to clinical management which presented an under 20x coverage were sequenced by Sanger technology. This process detects >99.5% of the replacement variants, of small insertions and deletions and CNV (duplications/deletions) of the LDLR gene.
Data were analysed with the Gendicall biocomputerised tool developed and validated by Gencardio and Genincode. The reads, with prior elimination of low quality extremes, were aligned against the genome grch37/hg19 version using the BWA-men programme. Detection of variants was made with the SAMtools and Gendicall programmes. Recording of the variants was made according to the HGVS standard using isoforms from the REFSEQ base of the Ensembl 81 repository. To aid variant classification protein function predictors were used (PolyPhen2, Mutation Taster and Provean) and splicing (MaxEntScan, FSPLICE, GeneSplicer and NNSplice). The interpretation and classification of pathogenicity/benign nature of variants was made in keeping with the information contained in the Gendiag.exe genetic variants database, following the regulations published by the American College of Medical Genetics and Genomics (ACMG). The variants classified as benign or possibly benign were not informed.10
Biochemical determinationsThe determination of total cholesterol, lipoprotein, cholesterol linked to high density lipoproteines (HDLc) LDLc apolipoprotein B and triglycerides in serum was made using enzymatic methods in the Cobas 8000 (Roche Diagnostics) modular analyser. The LDLc was determined with the Friedewald formula when the levels of triglycerides were under 400mg/dl.
Statistical analysisFirstly a univariate descriptive analysis of clinical and biochemical variables was undertaken for the description of these variables in the sample, according to the result of its genetic test. For bivariate analysis, a non parametric hypothesis contrast test was used for quantitative variables (Man-Witney U test).
In all cases a statistical significance of 5% (p<0.5) was required. Statistical analysis was performed using the statistical SPSS programme, version 21.0, with support from several calculus complements provided by the Microsoft Excel programme.
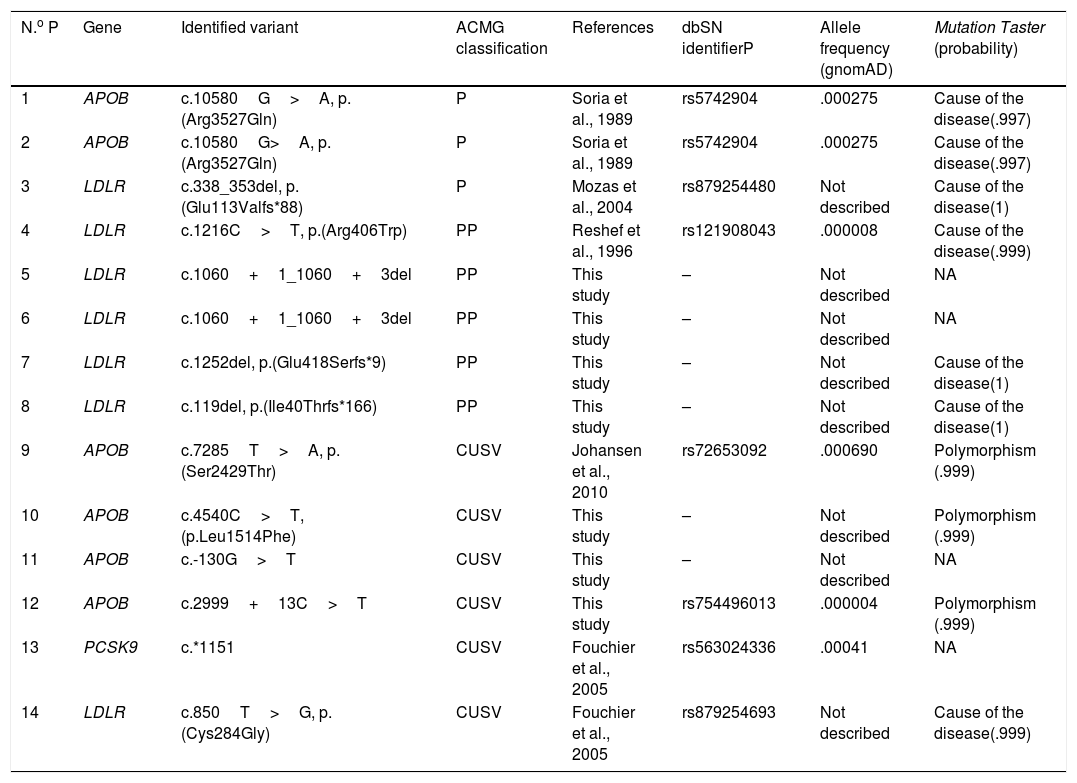
ResultsPatients p type of existing kinship; 8 positive test results, 2 with heterozygote pathogenic variants in the APOB gene (variant identified: p.(Arg3527Gln)), one with heterozygote pathogenic variant with the LDLR gene (identified variant: p.(Glu113Valfs*88)) and 5 with possibly pathogenic vagrants (variants identified : p.(Arg406Trp), c.1060+1_1060+3del, p.(Glu418Serfs*9), p.(Ile40Thrfs*166)).
Of the 5 variants identified in the gene LDLR, 3 are new variants, previously described in the literature (c.1060+1_1060+3del, p.(Glu418Serfs*9), p.(Ile40Thrfs*166)). Also, in 6 cases variants of uncertain clinical significance were detected in heterozygosis. The results of the genetic test practised are contained in Fig. 1 and in Table 1.11,12

Algorithm of patients assessed and results of the genetic test performed on the patients with ≥6 points on the Dutch Lipid Clinic Network.
CA: corneal arch; FHEIC: familial history of early ischaemic cardiopathy; APOB: apolipoprotein B; DLCN: Dutch Lipid Clinic Network; CVD: cardiovascular disease; FH: familial hypercholesterolaemia; HTG: hypertriglyceridaemia; LDLR: LDL receptor; PCSK9: proprotein convertase subtilisin/kexin type 9.
Genetic study findings using the Lipid InCode®test.
| N.o P | Gene | Identified variant | ACMG classification | References | dbSN identifierP | Allele frequency (gnomAD) | Mutation Taster (probability) |
|---|---|---|---|---|---|---|---|
| 1 | APOB | c.10580G>A, p.(Arg3527Gln) | P | Soria et al., 1989 | rs5742904 | .000275 | Cause of the disease(.997) |
| 2 | APOB | c.10580G>A, p.(Arg3527Gln) | P | Soria et al., 1989 | rs5742904 | .000275 | Cause of the disease(.997) |
| 3 | LDLR | c.338_353del, p.(Glu113Valfs*88) | P | Mozas et al., 2004 | rs879254480 | Not described | Cause of the disease(1) |
| 4 | LDLR | c.1216C>T, p.(Arg406Trp) | PP | Reshef et al., 1996 | rs121908043 | .000008 | Cause of the disease(.999) |
| 5 | LDLR | c.1060+1_1060+3del | PP | This study | – | Not described | NA |
| 6 | LDLR | c.1060+1_1060+3del | PP | This study | – | Not described | NA |
| 7 | LDLR | c.1252del, p.(Glu418Serfs*9) | PP | This study | – | Not described | Cause of the disease(1) |
| 8 | LDLR | c.119del, p.(Ile40Thrfs*166) | PP | This study | – | Not described | Cause of the disease(1) |
| 9 | APOB | c.7285T>A, p.(Ser2429Thr) | CUSV | Johansen et al., 2010 | rs72653092 | .000690 | Polymorphism (.999) |
| 10 | APOB | c.4540C>T, (p.Leu1514Phe) | CUSV | This study | – | Not described | Polymorphism (.999) |
| 11 | APOB | c.-130G>T | CUSV | This study | – | Not described | NA |
| 12 | APOB | c.2999+13C>T | CUSV | This study | rs754496013 | .000004 | Polymorphism (.999) |
| 13 | PCSK9 | c.*1151 | CUSV | Fouchier et al., 2005 | rs563024336 | .00041 | NA |
| 14 | LDLR | c.850T>G, p.(Cys284Gly) | CUSV | Fouchier et al., 2005 | rs879254693 | Not described | Cause of the disease(.999) |
ACMG: American College of Medical Genetics; APOB: Apolipoprotein B; dbSNP: Single Nucleotide Polymorphism Database; CUSV: clinically uncertain significance variant; gnomAD: The Genome Aggregation Database; LDLR: LDL receptor; NA: not available; P: pathogenic; PCSK9: proprotein convertase subtilisin/kexin type 9; PP: possibly pathogenic.
Of the patients included, we described those who had presented with genetic positivity to a mutation described as pathogenic for FH.
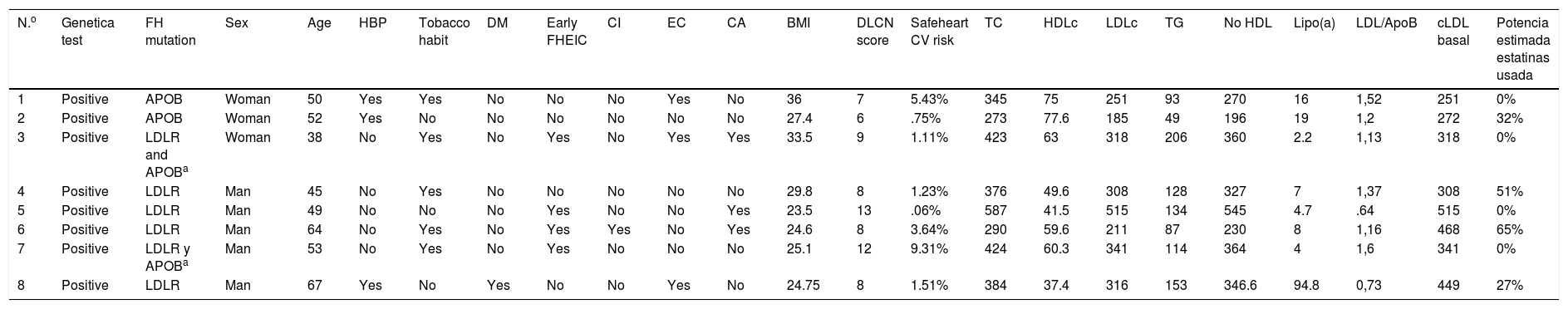
With regard to clinical characteristics, no major differences were observed for the presented mutation, just as for the parameters of the lipid profile on diagnosis. However, the patients presented with different cardiovascular risk factors at 5 years (Safeheart risk equation13) which probably were determined by other added factors, such as overweight, a tobacco habit, high blood pressure and control of LDLc levels (Tables 2 and 3). One of them, in their fifties, presented with a very high carotid disease and cardiovascular risk after 5 years, mostly due to other associated risk factors such as a tobacco habit and high blood pressure.
Clinical and analytical characteristics of patients who underwent a genetic test with a positive result to a pathogenic mutation in heterozygosis for familial hypercholesterolaemia.
| N.o | Genetica test | FH mutation | Sex | Age | HBP | Tobacco habit | DM | Early FHEIC | CI | EC | CA | BMI | DLCN score | Safeheart CV risk | TC | HDLc | LDLc | TG | No HDL | Lipo(a) | LDL/ApoB | cLDL basal | Potencia estimada estatinas usada |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Positive | APOB | Woman | 50 | Yes | Yes | No | No | No | Yes | No | 36 | 7 | 5.43% | 345 | 75 | 251 | 93 | 270 | 16 | 1,52 | 251 | 0% |
| 2 | Positive | APOB | Woman | 52 | Yes | No | No | No | No | No | No | 27.4 | 6 | .75% | 273 | 77.6 | 185 | 49 | 196 | 19 | 1,2 | 272 | 32% |
| 3 | Positive | LDLR and APOBa | Woman | 38 | No | Yes | No | Yes | No | Yes | Yes | 33.5 | 9 | 1.11% | 423 | 63 | 318 | 206 | 360 | 2.2 | 1,13 | 318 | 0% |
| 4 | Positive | LDLR | Man | 45 | No | Yes | No | No | No | No | No | 29.8 | 8 | 1.23% | 376 | 49.6 | 308 | 128 | 327 | 7 | 1,37 | 308 | 51% |
| 5 | Positive | LDLR | Man | 49 | No | No | No | Yes | No | No | Yes | 23.5 | 13 | .06% | 587 | 41.5 | 515 | 134 | 545 | 4.7 | .64 | 515 | 0% |
| 6 | Positive | LDLR | Man | 64 | No | Yes | No | Yes | Yes | No | Yes | 24.6 | 8 | 3.64% | 290 | 59.6 | 211 | 87 | 230 | 8 | 1,16 | 468 | 65% |
| 7 | Positive | LDLR y APOBa | Man | 53 | No | Yes | No | Yes | No | No | No | 25.1 | 12 | 9.31% | 424 | 60.3 | 341 | 114 | 364 | 4 | 1,6 | 341 | 0% |
| 8 | Positive | LDLR | Man | 67 | Yes | No | Yes | No | No | Yes | No | 24.75 | 8 | 1.51% | 384 | 37.4 | 316 | 153 | 346.6 | 94.8 | 0,73 | 449 | 27% |
APOB: apolipoprotein B; BMI: body mass index; CA: corneal arch; CD: carotid disease; CV: cardiovascular; DLCN: Dutch Lipid Clinic Network; DM diabetes mellitus; FH: familial hypercholesterolaemia; FHEIC: familial history of early ischaemic cardiopathy; HBP: high blood pressure; HDLc high-density lipoprotein cholesterol; IC: ischaemic cardiopathy; LDLc: low-density lipoprotein cholesterol; LDLR: LDL receptor; TC: total cholesterol, Lipo (a): lipoprotein a; TG: triglycerides.
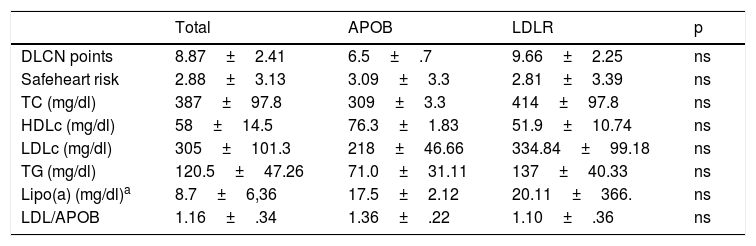
Comparative summary of levels of lipid profile of the patients according to type of mutation for FH they present.
| Total | APOB | LDLR | p | |
|---|---|---|---|---|
| DLCN points | 8.87±2.41 | 6.5±.7 | 9.66±2.25 | ns |
| Safeheart risk | 2.88±3.13 | 3.09±3.3 | 2.81±3.39 | ns |
| TC (mg/dl) | 387±97.8 | 309±3.3 | 414±97.8 | ns |
| HDLc (mg/dl) | 58±14.5 | 76.3±1.83 | 51.9±10.74 | ns |
| LDLc (mg/dl) | 305±101.3 | 218±46.66 | 334.84±99.18 | ns |
| TG (mg/dl) | 120.5±47.26 | 71.0±31.11 | 137±40.33 | ns |
| Lipo(a) (mg/dl)a | 8.7±6,36 | 17.5±2.12 | 20.11±366. | ns |
| LDL/APOB | 1.16±.34 | 1.36±.22 | 1.10±.36 | ns |
APOB: apolipoprotein B; DLCN: Dutch Lipid Clinic Network; FH: familial hypercholesterolaemia; HDLc high-density lipoprotein cholesterol; LCLR: LDL receptor; LDLc: low-density lipoprotein cholesterol; Lipo (a); lipoprotein a; TC: total cholesterol; TG: triglycerides.
It is also of note that there was an absence of a family background of early infarction in patients with APOB gene mutation compared with those who presented with the LDLR mutation where this was more prevalent. This could indicate better evolution of these patients with hypolipidemiant treatment.
Regarding treatment, the 2 patients with APOB mutations required current treatment with PCSK9 inhibitors although they presented with a sub-optimum control of LDLc levels. In the group with mutations of the LDLR gene, 5 patients were in treatment with this therapeutic group with an excellent control of LDLc levels (mean value of 32±12mg/dl).
DiscussionThe distribution of the genetic variants show a clear prevalence of the impact of the LDLR gene compared with the other alterations. However, given that according to the described frequency up until now in Spain, the mutations found for APOB were predominantly local to the north east area of the Iberian Peninsula, the fact that in this not very long period of time 2 families were found which were not linked to positivity of this mutation leads to the suspicion that there is a possibility of finding this somewhat more prevalent in the South East region of Spain, and that this finding is not anecdotal.
From a historical viewpoint, we found a possible explanation. The interrelationship between groups of humans from the north of the peninsula and the area around Huelva has 2 important moments in history. The first of them takes place during the second half of the first millennium BCE, when in the mountains are of Huelva new population settlements sprung up in conjunction with those of the plateau and the Duero basin, the celtici. Later literary sources bear witness to them, with the establishment of the Baeturia.14 The second of these moments takes place with the repopulation process of what is known as the “Banda gallega”· after the conquest of the region by Fernando III, in the 13th century. This event, which was nothing of a rarity, should be understood as the natural consequence of being immersed in the Silver Route, which acted as a communication force, linking both areas militarily and commercially.15
Thus, the transmission of this mutation throughout these centuries can be explained because it is an inherited pathology with implications in mortality but at later ages than procreation and that its penetration is not 100%. It remained thus generation after generation.
With regard to the prognostic of patients with pathogenic mutations in APOB for FH, a later presentation of heart disease has been described above 50 years of age and a better response to hypolipidemiant treatment, compared with patients who were affected by localised mutations in the LDLR gene. This appears to be the case in our group, although the risk of developing early atherosclerotic disease seems to be largely influenced by the amount of genetic impact, life habits and associated risk factors.
The 2 subjects with APOB mutation were in treatment with iPSCK9. However, control of LDLc levels was sub-optimum, unlike the group with the LDLR mutation where the therapeutic response was notable. One of them could be due to poor treatment compliance, in addition to dietary digression. In the other subject, despite correct administration of iPCSK9 the LDLc level did not change at all. This could be due to the mutation carried by these individuals, who generated a non functional LDLR, whereby circulating cholesterol did not pass into the inside of the hepatocyte.
In this cases series, the main limiting factor was the sample size which did not allow us to generate conclusions with greater evidence, but did allow us to discover more about this pathology, the true prevalence of which is as of yet unknown in our region.
ConclusionsAlthough the most common mutation in our sample was involvement of the LDLR gene, like in most populations, these findings could correspond to indications of a higher prevalence of pathogenic variants of the APOB gene in the southeast of the Iberian Peninsula to those known up until this time. We found historical facts, such as the presence of Celtic settlements and/or repopulations originating from Galicia centuries ago, which would offer a possible explanation of this and support this hypothesis.
FinancingThis study was financed with a grant from the Spanish Atherosclerosis Society (SEA for its initials in Spanish).
Conflict of interestThe authors have no conflict of interest to declare.
Please cite this article as: Roa Garrido J, Carrasco Salas P, Toscano Pérez C, Arrobas Velilla T, Vázquez Rico I, Díaz Fernández JF. Particularidades genéticas y bioquímicas de la hipercolesterolemia familiar en el suroeste de la Península Ibérica. Clin Investig Arterioscler. 2021;33:62–69.