




Hasta el momento, la mayor parte de los casos de hipercolesterolemia familiar (60-80%) se atribuyen a variantes patogénicas en el gen LDLR. Solo un 1-5% de los casos se produce por variantes en el gen APOB y un 0-3% por variantes en el gen PCSK9. Existen gran variedad en mutaciones patogénicas conocidas del gen LDLR mientras que, para las que afectan al gen APOB, la de mayor incidencia es p.Arg3527Gln, descrita predominantemente en poblaciones de Centroeuropa y América del Norte. En la Península Ibérica el gen predominante afectado es el del receptor de LDL, similar al resto del mundo, siendo la afectación del gen APOB descrita en individuos del noroeste y anecdótica en el resto del territorio. Analizamos genéticamente la población asistida en el primer año de una consulta de lípidos del suroeste de España con puntuación≥6 puntos de las clínicas de lípidos holandesas y describimos los hallazgos genéticos, bioquímicos y clínicos. Los primeros hallazgos muestran indicios de una posible mayor prevalencia de pacientes con mutación en el gen APOB respecto a otros territorios. Encontramos hechos históricos que darían una posible explicación a este hecho, apoyando así dicha presunción.
So far, most cases of hypercholesterolaemia (60-80%) are attributed to pathogenic variants in the LDLR gene. Only 1-5% of cases are caused by variants in the APOB gene, and 0-3% by variants in the PCSK9 gene. There is a large variety in known pathogenic mutations of the LDLR gene, while for those affecting the APOB gene, the highest incidence is p.Arg3527Gln, described predominantly in Central European and North American populations. In the Iberian Peninsula the predominant gene affected is that of the LDL receptor, similar to the rest of the world, with the involvement of the APOB gene being described in individuals from the northwest, and anecdotal in the rest of the territory. A genetics analysis was performed on the population attending the first year of a lipid clinic in southwestern Spain with a 6-point score from the Dutch lipid clinics. The genetic, biochemical and clinical findings are described. The first findings show indications of a possible higher prevalence of patients with mutation in the APOB gene compared to other territories. Historical evidence is presented that could give a possible explanation to this, thus supporting the assumption.
La hipercolesterolemia familiar (HF) es un trastorno genético común caracterizado por colesterol ligado a lipoproteínas de baja densidad (cLDL) circulante elevado, xantomas de tendones y enfermedad coronaria prematura. Se trata de la enfermedad genética más frecuente, con una prevalencia estimada de aproximadamente 1:250 según las últimas revisones1. Tiene un modo de herencia autosómico dominante y se produce por la presencia de variantes patogénicas en genes implicados en el metabolismo del receptor de LDL (LDLR)2. En la mayoría de los estudios, la frecuencia de mutaciones detectables en pacientes con HF heterocigota definida clínicamente o probable (puntuación de la red de clínicas de lípidos holandesas≥6 puntos) solo es del 60-80%, lo que indica que una proporción considerable de los pacientes con HF tienen una causa poligénica o la enfermedad está causada por genes aún por identificar2. En el 95% de los casos en los que se identifica una alteración genética patogénica, se localiza en el gen LDLR. En el resto de los estudios genéticos positivos, encontramos un 4-5% de los mismos con mutaciones en APOB y un 1% por mutaciones en el gen que codifica la proproteína convertasa de subtilisina/kexina tipo 9 (PCSK9), una proteína que induce la degradación del LDLR.
Posteriormente, se han propuesto otros 2 genes minoritarios adicionales como causantes de HF. Por un lado, se describió en 2014 mutaciones que afectan al gen STAP1 que codifican el adaptador de transducción de señal miembro de la familia 13. Sin embargo, recientemente Lamiquiz-Moneo et al.4 lo han puesto en entredicho demostrado que no parece presentar un papel etiológico por existir una ausencia de correlación entre genotipo y fenotipo. Por otro lado, Cenarro et al.5 describieron la mutación p.(Leu167del) en el gen APOE como la causa de HF autosómica dominante en el 3,1% de los sujetos que no presentaban mutación en otros genes ya conocidos, como son el LDLR, APOB ni PCSK9.
La HF presenta heterogenicidad genética puesto que se han descrito un número muy alto de variantes causales:>2.000 en LDLR; 32 en APOB, 23 en PCSK9 y 1 en APOE3.
La mutación más común encontrada en el gen APOB es una sustitución de glutamina por arginina en la posición 3527 (p.Arg3527Gln), que muestra deterioro de la capacidad de unión al LDLR y disminución de la captación de LDL.
La mutación p.Arg3527Gln se ha detectado casi exclusivamente en individuos caucásicos (frecuencia en población de ascendencia europea 1:500 a 1:700 individuos)6. En los pacientes inicialmente estudiados de Europa del Este y América del Norte, demostraron los mismos haplotipos del locus de ApoB. Su origen estaría situado en antepasados comunes que daten de dicha fecha en el periodo mesolítico (hace 10.000-6.000 años). Esta coincidencia de haplotipos se repite en la mayoría de los sujetos con dicha mutación alrededor del mundo con un origen en centroeuropa y con Los Alpes y Los Pirineos como barreras geográficas para la migración, presentando una baja incidencia en los pueblos mediterráneos. Además, dicha mutación estaba ya presente en los ancestros del periodo prehistórico del mesolítico en los pueblos celtas (Helvetii) que dominaban el territorio que se extendía desde el alto Rin y hasta el Meno y que emigraron al norte de la actual Suiza hace más de 2.000 años6,7.
Con relación a los datos conocidos en la Península Ibérica, se estima que la prevalencia de sujetos hipercolesterolémicos con un defecto familiar de apolipoproteína B en la población española general es muy baja (2,8×10-5). La mayoría de los casos (84%) se encuentran localizados en una región de ascendencia celta en el noroeste de España6.
El espectro de mutaciones correspondientes a la afectación de APOB ha aumentado en los últimos años, debido entre otros aspectos a las técnicas de secuenciación de próxima generación8. Recientemente, han sido estudiadas más de 300 variantes asociadas a HF pero solo el 6% de las mismas corresponden a APOB. Se sabe igualmente que las mutaciones en el gen APOB no tienen una penetrancia del 100% y que el fenotipo de los pacientes suele ser más leve que en pacientes con HF debido a mutaciones LDLR.
La mayoría de los heterocigotos con afectación del gen APOB responden bien a los medicamentos que reducen los niveles plasmáticos de LDL al inducir la actividad del receptor. También es conocido que raramente asocia enfermedad coronaria por debajo de los 50 años, a diferencia de los casos con otras variantes que afectan al LDLR6.
Analizamos las particularidades genéticas de la población asistida en la unidad de lípidos del suroeste de la Península Ibérica y su relación con las particularidades históricas regionales, junto a sus hallazgos clínicos y analíticos.
Material y métodosPacientesSe estableció un proyecto Arian nacional español para screening de HF según parámetros de laboratorio (LDL≥250mg/dl), del cual nuestro centro fue partícipe. Han sido analizados pacientes desde marzo de 2018 hasta marzo de 2019.
En primer lugar, se descartaron aquellos pacientes con causas secundarias de dislipidemia, como son el hipotiroidismo, síndrome nefrótico, enfermedad renal avanzada, colestasis hepática, consumo excesivo de alcohol, embarazo y fármacos (antirretrovirales, anabolizantes, estrógenos, progestágenos, glucocorticoides, tacrolimus y ciclosporina). Se les realizaba una anotación para volver a ser evaluados en caso de persistencia elevada de cifras de cLDL tras la resolución de la causa secundaria. Los pacientes que presentaban diabetes fueron evaluados en consulta puesto que presentan muy alto riesgo cardiovascular, además de poder coexistir dicho diagnóstico con HF.
Los restantes fueron valorados en consulta, donde se realizó una historia clínica completa recogiéndose factores de riesgo cardiovasculares clásicos (hipertensión arterial, diabetes, tabaquismo, etc.) y screening de enfermedad cardiovascular, clínica y subclínica. Se aplicaron los criterios de la red de clínicas de lípidos holandesas (Dutch Lipid Clinic Network)9 y a aquellos con una puntuación≥6 puntos, y por tanto con diagnóstico probable o cierto para HF, se les practicó el estudio genético, previa obtención del consentimiento informado.
El origen geográfico de los pacientes corresponde a nuestro centro hospitalario, situado en suroeste de España, tratándose de sujetos no relacionados de diferentes familias.
Se extrajeron muestras de sangre total para estudios de ADN y de suero para la determinación de los parámetros bioquímicos.
Extracción de ADNLa extracción de ADN se realizó a partir de 5ml de sangre periférica anticoagulada con EDTA, usando un kit comercial (Puregene: DNA kit de aislamiento, Gentra Systems, MN. EE. UU.).
Panel de secuenciación de próxima generaciónSe realizó el panel Lipid inCode® (Novartis®) en el laboratorio concertado Lipid in Code (La Coruña, España). Este panel permite analizar las secuencias de ADN promotoras, codificantes y regiones de unión exón-intrón (±25pb) de los genes LDLR (NM_000527.4), APOB (NM_000384.2), PCSK9 (NM_174936.3), APOE (NM_000041.3), STAP1 (NM_012108.3;), LDLRAP1 (NM_015627.2) y LIPA (NM_000235.3).
El enriquecimiento de la muestra en las regiones de interés se llevó a cabo mediante la química SureSelect QXT de Agilent (solución de enriquecimiento diseñada y validada por Gencardio/GenIncode). Las librerías resultantes se secuenciaron de manera masiva en la plataforma de Illumina MiSeq System, utilizando química basada en amplificación clonal y secuenciación por síntesis. Las regiones génicas con call rate<100% a una cobertura de 30x así como las variantes asociadas al manejo clínico que presentaron una cobertura inferior a 20x, se secuenciaron por tecnología Sanger. Este proceso detecta>99,5% de las variantes de sustitución, de las pequeñas inserciones y deleciones y los CNV (duplicaciones/deleciones) del gen LDLR.
Los datos se analizan con la herramienta bioinformática Gendicall desarrollada y validada por Gencardio y Genincode. Los reads, previa eliminación de los extremos de baja calidad, son alineados contra la versión del genoma grch37/hg19 mediante el programa BWA-men. La detección de variantes se realiza con los programas SAMtools y Gendicall. La anotación de las variantes se realizó según el estándar HGVS usando isoformas de la base REFSEQ del repositorio de Ensembl 81. Se han utilizado para ayudar a la clasificación de variantes los predictores de función de proteína (PolyPhen2, Mutation Taster y Provean) y de splicing (MaxEntScan, FSPLICE, GeneSplicer y NNSplice). La interpretación y clasificación de patogenicidad/benignidad de las variantes se ha realizado conforme a la información contenida en la base de datos de variantes genéticas de Gendiag.exe, siguiendo las reglas publicadas por el American Collegue of Medical Genetics and Genomics (ACMG). Las variantes clasificadas como benignas o posiblemente benignas no son informadas10.
Determinaciones bioquímicasSe realizó la determinación en suero de colesterol total, lipoproteína a, colesterol ligado a lipoproteínas de alta densidad (cHDL), cLDL apolipoproteína B y triglicéridos mediante métodos enzimáticos en el analizador modular Cobas 8000 (Roche Diagnostics). El cLDL se determinó con la fórmula de Friedewald cuando los niveles de triglicéridos fueron menores de 400mg/dl.
Análisis estadísticoEn primer lugar se llevará a cabo un análisis descriptivo univariado de variables clínicas y bioquímicas para la descripción de estas variables en la muestra según resultado de su test genético. Para el análisis bivariado, se recurrirá a una prueba de contraste de hipótesis no paramétricas para variables cuantitativas (U Man-Witney).
En todos los casos se exige una significación estadística del 5% (p<0,05). Los análisis estadísticos se realizaron mediante el programa estadístico SPSS, en su versión 21.0, con el apoyo de ciertos complementos de cálculo proporcionado por el programa Microsoft Excel.
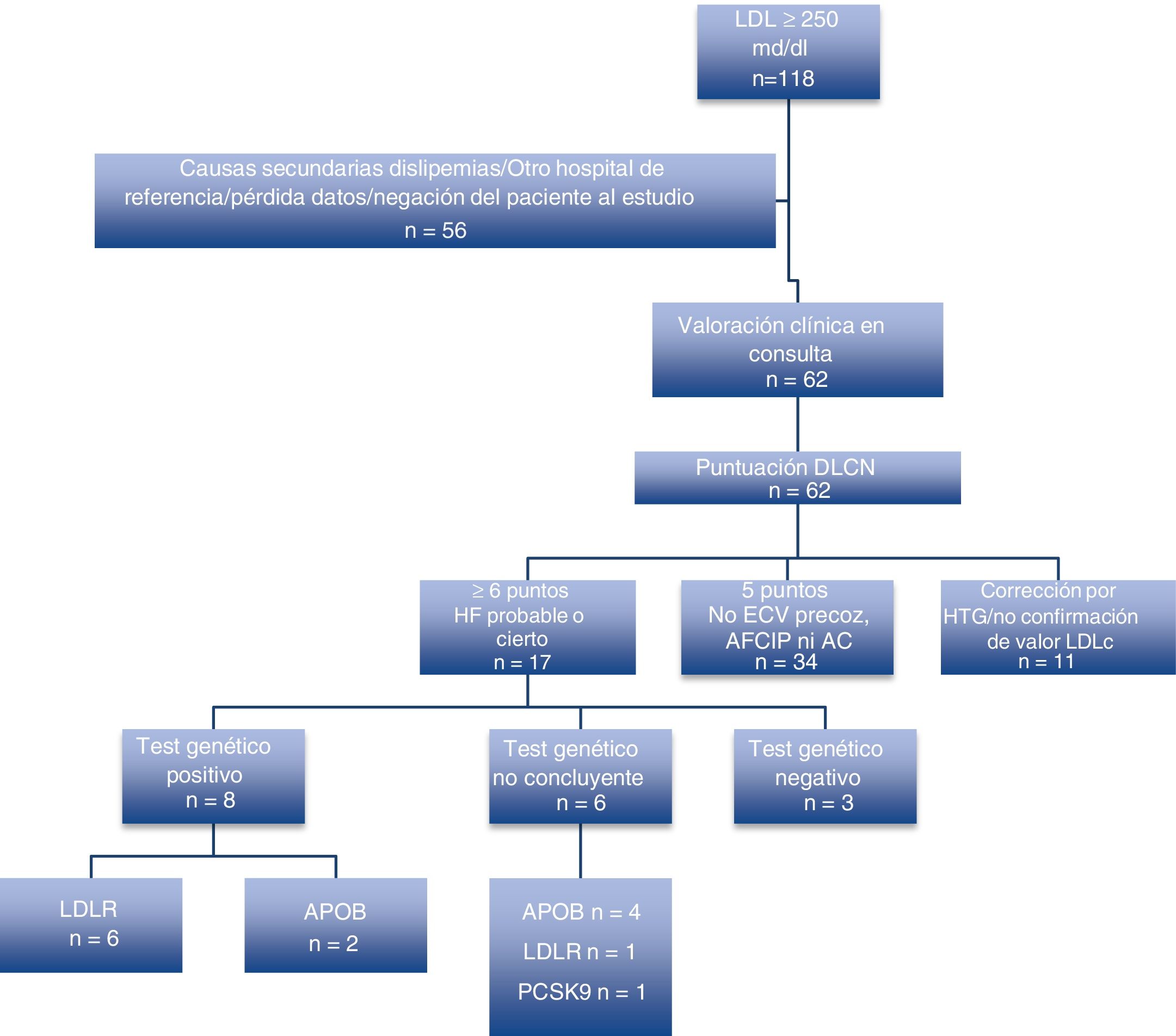
ResultadosLos pacientes que se describen son los que han presentado positividad genética a una mutación descrita como patogénica para la HF. Durante el primer año de consulta, del total de 118 iniciales analizados por LDL elevadas, fueron finalmente vistos en consulta 62, siendo el resto descartados por causas secundarias o por pertenecer a otras áreas sanitarias.
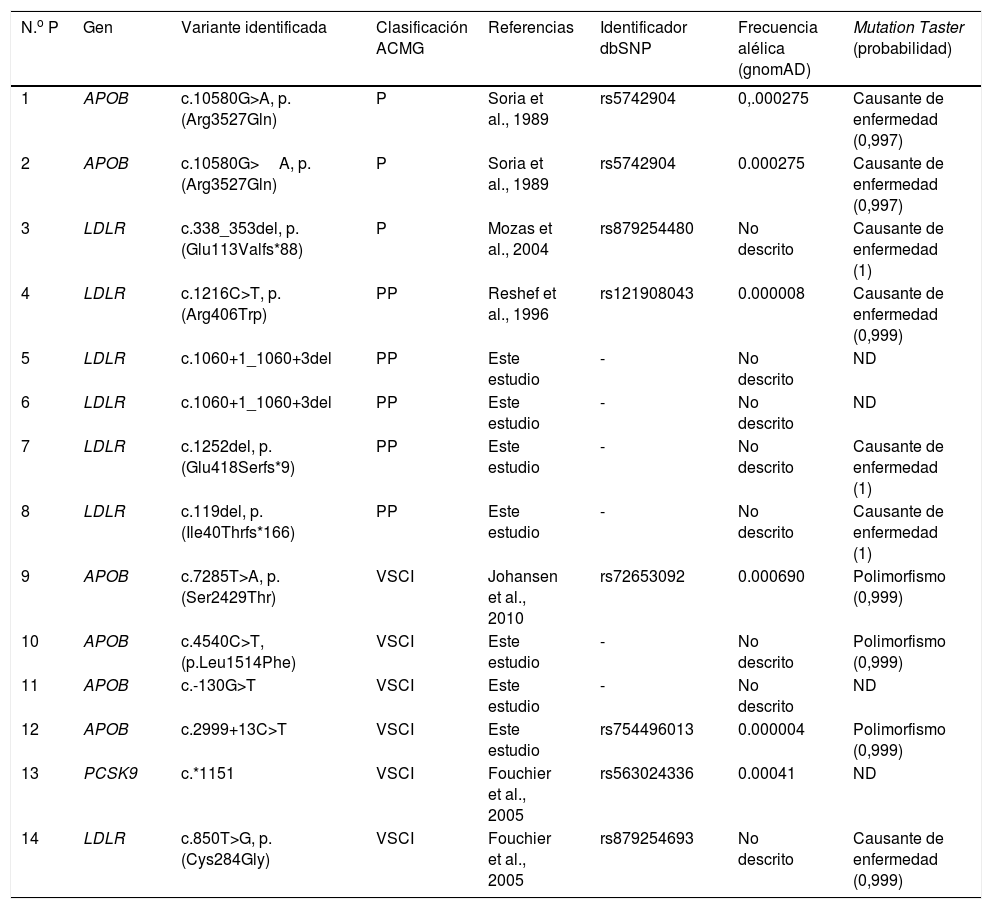
Se solicitaron un total de 17 test genéticos para HF por presentar una puntación≥6 puntos de la red de clínicas de lípidos holandesas de pacientes no relacionados sin existir ningún tipo de parentesco; resultado un total de 8 test positivos, 2 con variantes patogénicas en heterocigosis en el gen APOB (variante identificada p.(Arg3527Gln)), uno con variante patogénica heterocigota en el gen LDLR (variante identificada: p.(Glu113Valfs*88)) y 5 con variantes posiblemente patogénicas (variantes identificadas: p.(Arg406Trp), c.1060+1_1060+3del, p.(Glu418Serfs*9), p.(Ile40Thrfs*166)).
De las 5 variantes identificadas en el gen LDLR, 3 son variantes nuevas, no descritas previamente en la literatura (c.1060+1_1060+3del, p.(Glu418Serfs*9), p.(Ile40Thrfs*166)). Además, en 6 casos se detectaron variantes de significado clínico incierto en heterocigosis. Los resultados de los test genéticos practicados quedan recogidos en la figura 1 y en la tabla 111-12.

Algoritmo de pacientes valorados y resultados de test genéticos realizados a los pacientes con≥6 puntos de las Clínicas de Lípidos Holandesas. AC: arco corneal; AFCIP: antecedente familiar de cardiopatía isquémica precoz; APOB: apolipoproteína B; DLCN: Dutch Lipid Clinic Network; ECV: enfermedad cardiovascular; HF: hipercolesterolemia familiar; HTG: hipertrigliceridemia; LDLR: receptor de LDL; PCSK9: proproteína convertasa subtilisina/kexina tipo 9.
Hallazgos en el estudio genético mediante el test Lipid InCode®
| N.o P | Gen | Variante identificada | Clasificación ACMG | Referencias | Identificador dbSNP | Frecuencia alélica (gnomAD) | Mutation Taster (probabilidad) |
|---|---|---|---|---|---|---|---|
| 1 | APOB | c.10580G>A, p.(Arg3527Gln) | P | Soria et al., 1989 | rs5742904 | 0,.000275 | Causante de enfermedad (0,997) |
| 2 | APOB | c.10580G>A, p.(Arg3527Gln) | P | Soria et al., 1989 | rs5742904 | 0.000275 | Causante de enfermedad (0,997) |
| 3 | LDLR | c.338_353del, p.(Glu113Valfs*88) | P | Mozas et al., 2004 | rs879254480 | No descrito | Causante de enfermedad (1) |
| 4 | LDLR | c.1216C>T, p.(Arg406Trp) | PP | Reshef et al., 1996 | rs121908043 | 0.000008 | Causante de enfermedad (0,999) |
| 5 | LDLR | c.1060+1_1060+3del | PP | Este estudio | - | No descrito | ND |
| 6 | LDLR | c.1060+1_1060+3del | PP | Este estudio | - | No descrito | ND |
| 7 | LDLR | c.1252del, p.(Glu418Serfs*9) | PP | Este estudio | - | No descrito | Causante de enfermedad (1) |
| 8 | LDLR | c.119del, p.(Ile40Thrfs*166) | PP | Este estudio | - | No descrito | Causante de enfermedad (1) |
| 9 | APOB | c.7285T>A, p.(Ser2429Thr) | VSCI | Johansen et al., 2010 | rs72653092 | 0.000690 | Polimorfismo (0,999) |
| 10 | APOB | c.4540C>T, (p.Leu1514Phe) | VSCI | Este estudio | - | No descrito | Polimorfismo (0,999) |
| 11 | APOB | c.-130G>T | VSCI | Este estudio | - | No descrito | ND |
| 12 | APOB | c.2999+13C>T | VSCI | Este estudio | rs754496013 | 0.000004 | Polimorfismo (0,999) |
| 13 | PCSK9 | c.*1151 | VSCI | Fouchier et al., 2005 | rs563024336 | 0.00041 | ND |
| 14 | LDLR | c.850T>G, p.(Cys284Gly) | VSCI | Fouchier et al., 2005 | rs879254693 | No descrito | Causante de enfermedad (0,999) |
ACMG: American College of Medical Genetics; APOB: apolipoproteína B; dbSNP: Single Nucleotide Polymorphism Database; gnomAD: The Genome Aggregation Database; LDLR: receptor de LDL; ND: no disponible; P: patogénica; PCSK9: proproteína convertasa subtilisina/kexina tipo 9; PP: posiblemente patogénica; VSCI: variante de significado clínico incierto.
De los pacientes recogidos, describimos los que han presentado positividad genética a una mutación descrita como patogénica para la HF.
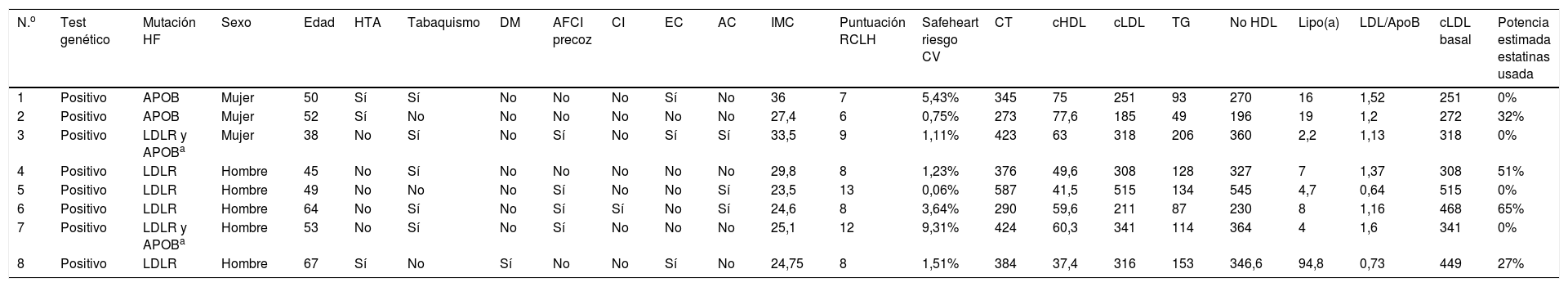
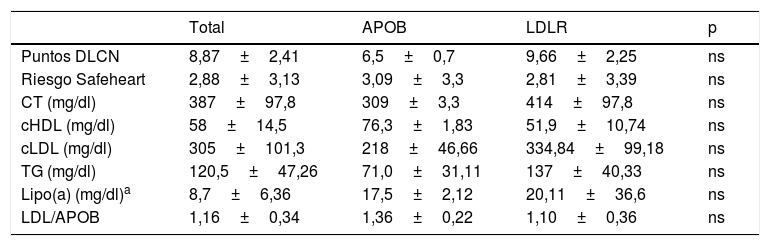
Con relación a las características clínicas, no se observan grandes diferencias según la mutación presentada, al igual que los parámetros del perfil lipídico en el momento del diagnóstico. No obstante, los pacientes presentan diferente perfil de riesgo cardiovascular a 5 años (Safeheart risk equation13) que probablemente viene determinado por otros factores añadidos, como son el sobrepeso, tabaquismo, hipertensión arterial o el control de las cifras de cLDL (tablas 2 y 3). Uno de ellos, en la quinta década de la vida, presenta enfermedad carotídea y riesgo cardiovascular a 5 años muy alto, debido en gran parte a otros factores de riesgo asociados como son el tabaquismo y la hipertensión arterial.
Características clínicas y analíticas de los pacientes sometidos a test genético con resultado positivo a mutación patogénica en heterocigosis para hipercolesterolemia familiar
| N.o | Test genético | Mutación HF | Sexo | Edad | HTA | Tabaquismo | DM | AFCI precoz | CI | EC | AC | IMC | Puntuación RCLH | Safeheart riesgo CV | CT | cHDL | cLDL | TG | No HDL | Lipo(a) | LDL/ApoB | cLDL basal | Potencia estimada estatinas usada |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Positivo | APOB | Mujer | 50 | Sí | Sí | No | No | No | Sí | No | 36 | 7 | 5,43% | 345 | 75 | 251 | 93 | 270 | 16 | 1,52 | 251 | 0% |
| 2 | Positivo | APOB | Mujer | 52 | Sí | No | No | No | No | No | No | 27,4 | 6 | 0,75% | 273 | 77,6 | 185 | 49 | 196 | 19 | 1,2 | 272 | 32% |
| 3 | Positivo | LDLR y APOBa | Mujer | 38 | No | Sí | No | Sí | No | Sí | Sí | 33,5 | 9 | 1,11% | 423 | 63 | 318 | 206 | 360 | 2,2 | 1,13 | 318 | 0% |
| 4 | Positivo | LDLR | Hombre | 45 | No | Sí | No | No | No | No | No | 29,8 | 8 | 1,23% | 376 | 49,6 | 308 | 128 | 327 | 7 | 1,37 | 308 | 51% |
| 5 | Positivo | LDLR | Hombre | 49 | No | No | No | Sí | No | No | Sí | 23,5 | 13 | 0,06% | 587 | 41,5 | 515 | 134 | 545 | 4,7 | 0,64 | 515 | 0% |
| 6 | Positivo | LDLR | Hombre | 64 | No | Sí | No | Sí | Sí | No | Sí | 24,6 | 8 | 3,64% | 290 | 59,6 | 211 | 87 | 230 | 8 | 1,16 | 468 | 65% |
| 7 | Positivo | LDLR y APOBa | Hombre | 53 | No | Sí | No | Sí | No | No | No | 25,1 | 12 | 9,31% | 424 | 60,3 | 341 | 114 | 364 | 4 | 1,6 | 341 | 0% |
| 8 | Positivo | LDLR | Hombre | 67 | Sí | No | Sí | No | No | Sí | No | 24,75 | 8 | 1,51% | 384 | 37,4 | 316 | 153 | 346,6 | 94,8 | 0,73 | 449 | 27% |
AC: arco corneal; AFCI: antecedente familiar de cardiopatía isquémica; APOB: apolipoproteína B; cHDL: colesterol ligado a lipoproteínas de alta densidad [High density lipoprotein]; cLDL: colesterol ligado a lipoproteínas de baja densidad [Low density lipoprotein]; CI: cardiopatía isquémica; CT: colesterol total; CV: cardiovascular; DM: diabetes mellitus; EC: enfermedad carotídea; HF: hipercolesterolemia familiar; HTA: hipertensión arterial; IMC: índice de masa corporal; LDLR: receptor LDL; Lipo(a): lipoproteína a RCLH: Red de Clínicas de Lípidos Holandesas; TG: triglicéridos.
Resumen comparativo de los valores del perfil lipídico de los pacientes según el tipo de mutación para HF que presenten
| Total | APOB | LDLR | p | |
|---|---|---|---|---|
| Puntos DLCN | 8,87±2,41 | 6,5±0,7 | 9,66±2,25 | ns |
| Riesgo Safeheart | 2,88±3,13 | 3,09±3,3 | 2,81±3,39 | ns |
| CT (mg/dl) | 387±97,8 | 309±3,3 | 414±97,8 | ns |
| cHDL (mg/dl) | 58±14,5 | 76,3±1,83 | 51,9±10,74 | ns |
| cLDL (mg/dl) | 305±101,3 | 218±46,66 | 334,84±99,18 | ns |
| TG (mg/dl) | 120,5±47,26 | 71,0±31,11 | 137±40,33 | ns |
| Lipo(a) (mg/dl)a | 8,7±6,36 | 17,5±2,12 | 20,11±36,6 | ns |
| LDL/APOB | 1,16±0,34 | 1,36±0,22 | 1,10±0,36 | ns |
APOB: apolipoproteína B; cHDL: colesterol ligado a lipoproteínas de alta densidad [High density lipoprotein]; cLDL: colesterol ligado a lipoproteínas de baja densidad [Low density lipoprotein]; CT: colesterol total; DLCN: Dutch Lipid Clinic Network; HF: hipercolesterolemia familiar; LDLR: receptor LDL; Lipo(a): lipoproteína a; TG: triglicéridos.
Por otro lado, cabría destacar la ausencia de antecedentes familiares de infarto precoz en los pacientes con mutación en el gen APOB respecto a los que presentación mutación en LDLR entre los que es más prevalente, que podría indicar mejor evolución de estos pacientes con tratamiento hipolipidemiante.
Con relación al tratamiento, los 2 pacientes con mutaciones en APOB requieren actualmente tratamiento con inhibidores de PCSK9 aunque presentan un control no óptimo por el momento de cifras de cLDL. En el grupo con mutación en el gen LDLR, 5 pacientes se encuentran en tratamiento con este grupo terapéutico con excelente control de cifras de cLDL (valor medio 32±12mg/dl).
DiscusiónLa distribución de las variantes genéticas muestra una prevalencia clara de la afectación del gen LDLR frente al resto de alteraciones. No obstante, puesto que según la frecuencia descrita hasta el momento en nuestro país, las mutaciones encontradas para APOB han sido predominantemente localizadas a la zona noroeste de la Península Ibérica, el hecho de encontrar en este periodo de tiempo no muy largo a 2 familias no relacionadas con positividad a esta mutación hace sospechar la posibilidad de encontrarse de forma más prevalente en la región suroeste de España, no tratándose de un hallazgo anecdótico.
Desde un punto de vista histórico, encontramos una posible explicación. La interrelación entre grupos humanos procedentes del norte peninsular y el área onubense tiene 2 momentos importantes en la Historia. El primero de ellos tiene lugar durante la segunda mitad del i milenio a.C., cuando en la sierra de Huelva se dan nuevas ocupaciones de poblaciones emparentadas con las culturas de la meseta y la cuenca del Duero, los celtici de los que dan testimonio las posteriores fuentes literarias que dan lugar al concepto de Baeturia14. El segundo de estos momentos tiene lugar con el proceso de repoblación de la conocida como «Banda gallega», tras la conquista del lugar por Fernando III, en el s. xiii. Este hecho, lejos de considerarse una rareza, debe entenderse como la consecuencia natural de encontrarse inmersos en la Vía de la Plata, elemento de comunicación que había unido militar y comercialmente ambas zonas15.
Así, la trasmisión de esta mutación a lo largo de estos siglos puede ser explicada porque se trate de una patología heredada con implicaciones en la mortalidad pero a edades posteriores a la época de procreación y a que su penetrancia no es del 100%, permaneciendo así generación tras generación.
En lo referido al pronóstico de los pacientes con mutaciones patogénicas en APOB para HF, se ha descrito una aparición más tardía de enfermedad coronaria más allá de los 50 años y una mejor respuesta al tratamiento hipolipidemiante, frente a los pacientes afectos con mutaciones localizadas en el gen de LDLR. Este hecho parece observarse en nuestro grupo, aunque el riesgo de desarrollar enfermedad aterosclerótica de forma precoz parece estar influido en gran medida por la suma que conlleva la afectación genética y los hábitos de vida y factores de riesgo asociados.
Los 2 sujetos con mutación APOB se encuentran en tratamiento con iPSCK9. Sin embargo, el control de cifras de cLDL es subóptimo, a diferencia del grupo con mutación LDLR en los que la respuesta terapéutica ha sido notable. Uno de ellos podría ser debido a un mal cumplimiento terapéutico, además de digresión dietética. En el otro sujeto, a pesar de una correcta administración de iPCSK9 no ha modificado absolutamente en nada el valor de cLDL. Ello podría ser debido a la mutación que portan estos individuos, que generan un LDLR no funcional, por lo que el colesterol circulante no pasa al interior del hepatocito.
En esta serie de casos, el principal factor limitante es el tamaño de la muestra que no permite generar conclusiones con mayor evidencia, sin embargo, nos permite averiguar más sobre esta patología aún por conocer la verdadera prevalencia en nuestra región.
ConclusionesAunque la mutación más frecuente en nuestra muestra es la afectación del gen LDLR como en la mayoría de poblaciones, estos hallazgos podrían corresponder a indicios de una prevalencia de variantes patogénicas del gen de APOB más elevada en el suroeste de la Península Ibérica de lo conocido hasta el momento. Encontramos hechos históricos, como la presencia de pueblos celtas y/o repoblaciones procedentes de Galicia hace siglos, que darían una posible explicación a este hecho, apoyando así dicha presunción.
FinanciaciónEste trabajo ha sido financiado con una beca de la SEA (Sociedad Española de Aterosclerosis).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.