





Synovial sarcoma is a well-recognized soft tissue malignancy in adult population. Primary intraabdominal synovial sarcoma is a rare occurrence, of which 1% has retroperitoneal location. These are firm, rounded, slowly growing, well-circumscribed tumors which can sometimes be of multinodular texture. Histopathology and Immunohistochemistry (IHC) aid in confirming the diagnosis. Nearly 90% of the cases have an associated chromosomal translocation (X; 18), (p11; q11). Given its rare occurrence, management of this malignancy is challenging, though radical surgery combined with chemotherapy and radiotherapy have been described.
A 54-year-old, grand multiparous lady was referred to gynecologic oncology clinic due to a large pelvic mass and abnormal uterine bleeding. Pelvic MRI showed solid cystic pelvic mass in the left adnexa. She underwent laparotomy and bilateral salpingo-oophorectomy, hysterectomy, omentectomy and pelvic lymphadenectomy. Final pathology report revealed synovial sarcoma.
Synovial sarcoma should be considered in the differential diagnosis of abdomino-pelvic masses in young and middle-aged patients.
El sarcoma sinovial es una neoplasia maligna de tejidos blandos bien reconocida en la población adulta. El sarcoma sinovial intraabdominal primario es una ocurrencia rara, de la cual el 1% tiene localización retroperitoneal. Se trata de tumores firmes, redondeados, de crecimiento lento y bien delimitados, que en ocasiones pueden tener una textura multinodular. La histopatología y la inmunohistoquímica (IHC) ayudan a confirmar el diagnóstico. Casi el 90% de los casos tienen una translocación cromosómica asociada (X; 18), (p11; q11). Dada su rara ocurrencia, el manejo de esta neoplasia maligna es un desafío, aunque se ha descrito cirugía radical combinada con quimioterapia y radioterapia.
Una mujer adulta multípara de 54 años fue remitida a la clínica de oncología ginecológica debido a una gran masa pélvica y sangrado uterino anormal. La resonancia magnética pélvica mostró una masa pélvica quística sólida en el anexo izquierdo. Fue sometida a laparotomía y salpingooforectomía bilateral, histerectomía, omentectomía y linfadenectomía pélvica. El informe patológico final reveló sarcoma sinovial.
El sarcoma sinovial debe considerarse en el diagnóstico diferencial de masas abdominopélvicas en pacientes jóvenes y de mediana edad.
Synovial sarcoma (SS) is a well-recognized soft tissue malignancy particularly in young population, although no age is spared.1 It constitutes 2.5–10.5% of all primary soft tissue malignancies2 and prevalence of 7.25 per 100,000 is reported for the disease.3
Three subtypes are identified for this condition, based on histological findings: Biphasic, monophasic, and poorly differentiated. Twenty to thirty percent of synovial sarcomas are of biphasic type, and2 contain spindle cell component with varying proportions of epithelial tissue. The epithelial component can be of either glandular or solid textures. In the former, the glandular lumen is lined by cuboid or columnar cells and may contain epithelial mucin. The cellular nuclei are oval and vesicular and cells contain conspicuous eosinophilic cytoplasm, making them noticeable in the background of spindle cells.4
Postsurgical pathology and immunohistochemistry testing of the resected lesions aid in diagnosing synovial sarcoma.5
The condition has a known genetic etiology, namely the translocation (X; 18), (p11; q11), which is reported to be present in nearly 90% of patients with the diagnosis.3
Rarity and mild symptoms delay the precise diagnosis, resulting in general hospital visits, with inadequate therapy, and undesirable outcomes.
Primary abdominal synovial sarcoma is a rarity; herein we present a case of synovial sarcoma in a 54-year-old woman with a large pelvic mass.
Case presentationA 54-year-old grand multiparous lady was referred to our gynecologic oncology clinic due to large pelvic mass and abnormal uterine bleeding. She had history of 8 normal vaginal deliveries and 1 cesarian section.
She was diagnosed with diabetes mellitus one year ago. She was addicted to opium.
General physical exam was normal. Pelvic exam confirmed a pelvic mass suggestive of pedunculated myoma. On abdominal ultrasonography with Doppler, a 118×88mm pedunculated growth was noted near uterine body with vascular flow in some areas, indicating a left pelvic mass. Pelvic MRI, performed with and without intravenous contrast and reviewed by an expert radiologist, confirmed presence of a pelvic mass with mixed intensity (solid-cystic) suggesting left adnexal mass, probably of an ovarian origin. All but one of the tumor markers were normal, as follows: He4=167 (up to 150), and normal CA125, CEA, AFP AND beta HCG. She was candidate for surgery. Endometrial biopsy, pap smear, colposcopy and other preoperative tests were all normal. She underwent laparotomy via midline incision. Intraoperative findings revealed a left pelvic mass separate from the left ovary. Frozen section of the mass suggested adenocarcinoma originated from stomach/colon or an endometrioid ovarian carcinoma. Bilateral salpingo-oophorectomy, hysterectomy, omentectomy and pelvic lymphadenectomy was performed following peritoneal cytology. Final pathology report, diagnosed the mass as a synovial sarcoma of biphasic type (Fig. 1) while all the other specimens were reported to be normal. Postoperative expert panel suggested complementary X,18 Translocation. Study on remaining pelvic mass blocks was negative for SS18(18q11) rearrangement, but extra copy of SS18 was reported in about 27% of tumor cells, suggesting RT_PCR. On follow up review after twelve months, patient had no problems.
Discussion
One of the commonest sarcomas in adult population is the synovial sarcoma, nearly 30% of cases of which are diagnosed in the first two decades of life. The disease shows female preponderance, and can be manifested at any age, with the mean age at diagnosis reported to be 30 years3; our patient was 56 years old.
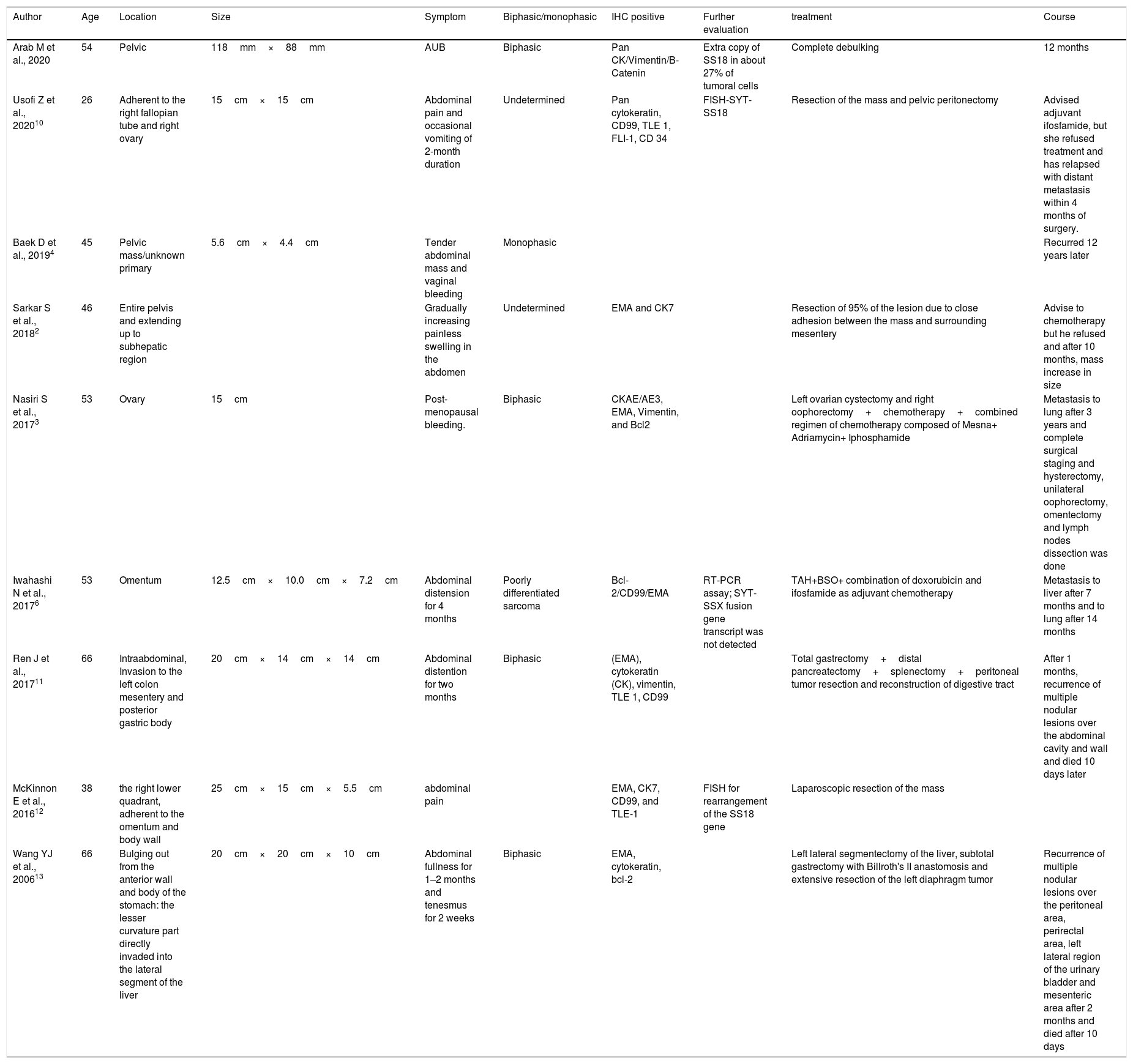
Synovia and soft tissues are not exclusively the sites of origin of synovial sarcomas. Nearly 70% of cases of synovial sarcoma occur in lower extremities,2 particularly the knee. Occurrence of the disease at other sites (e.g. lungs, kidneys, ovaries pelvis, pleural, and uterus) has been reported.6 In our patient's case, it was located in pelvic cavity. Primary intraabdominal synovial sarcoma is a rare entity, and less than 1% of primary synovial malignancies are located in retroperitoneum.2Table 1 summarizes the eight female patients with intra-abdominal SS, reported in literature. Synovial sarcomas are high-grade tumors, and their symptomatology depends on their location. Tumor site and the rate of its growth determines its gross appearance. The slowly growing masses are well-circumscribed, firm, round, or of a multinodular appearance; these masses may develop prominent cystic changes. On the other hand, masses with rapidly growth are poorly circumscribed lesions which look variegated and friable. Hemorrhagic and necrotic regions may develop in these tumors.2
The eight female patients with intra-abdominal SS reported in literature.
| Author | Age | Location | Size | Symptom | Biphasic/monophasic | IHC positive | Further evaluation | treatment | Course |
|---|---|---|---|---|---|---|---|---|---|
| Arab M et al., 2020 | 54 | Pelvic | 118mm×88mm | AUB | Biphasic | Pan CK/Vimentin/B-Catenin | Extra copy of SS18 in about 27% of tumoral cells | Complete debulking | 12 months |
| Usofi Z et al., 202010 | 26 | Adherent to the right fallopian tube and right ovary | 15cm×15cm | Abdominal pain and occasional vomiting of 2-month duration | Undetermined | Pan cytokeratin, CD99, TLE 1, FLI-1, CD 34 | FISH-SYT-SS18 | Resection of the mass and pelvic peritonectomy | Advised adjuvant ifosfamide, but she refused treatment and has relapsed with distant metastasis within 4 months of surgery. |
| Baek D et al., 20194 | 45 | Pelvic mass/unknown primary | 5.6cm×4.4cm | Tender abdominal mass and vaginal bleeding | Monophasic | Recurred 12 years later | |||
| Sarkar S et al., 20182 | 46 | Entire pelvis and extending up to subhepatic region | Gradually increasing painless swelling in the abdomen | Undetermined | EMA and CK7 | Resection of 95% of the lesion due to close adhesion between the mass and surrounding mesentery | Advise to chemotherapy but he refused and after 10 months, mass increase in size | ||
| Nasiri S et al., 20173 | 53 | Ovary | 15cm | Post-menopausal bleeding. | Biphasic | CKAE/AE3, EMA, Vimentin, and Bcl2 | Left ovarian cystectomy and right oophorectomy+chemotherapy+combined regimen of chemotherapy composed of Mesna+ Adriamycin+ Iphosphamide | Metastasis to lung after 3 years and complete surgical staging and hysterectomy, unilateral oophorectomy, omentectomy and lymph nodes dissection was done | |
| Iwahashi N et al., 20176 | 53 | Omentum | 12.5cm×10.0cm×7.2cm | Abdominal distension for 4 months | Poorly differentiated sarcoma | Bcl-2/CD99/EMA | RT-PCR assay; SYT-SSX fusion gene transcript was not detected | TAH+BSO+ combination of doxorubicin and ifosfamide as adjuvant chemotherapy | Metastasis to liver after 7 months and to lung after 14 months |
| Ren J et al., 201711 | 66 | Intraabdominal, Invasion to the left colon mesentery and posterior gastric body | 20cm×14cm×14cm | Abdominal distention for two months | Biphasic | (EMA), cytokeratin (CK), vimentin, TLE 1, CD99 | Total gastrectomy+distal pancreatectomy+splenectomy+peritoneal tumor resection and reconstruction of digestive tract | After 1 months, recurrence of multiple nodular lesions over the abdominal cavity and wall and died 10 days later | |
| McKinnon E et al., 201612 | 38 | the right lower quadrant, adherent to the omentum and body wall | 25cm×15cm×5.5cm | abdominal pain | EMA, CK7, CD99, and TLE-1 | FISH for rearrangement of the SS18 gene | Laparoscopic resection of the mass | ||
| Wang YJ et al., 200613 | 66 | Bulging out from the anterior wall and body of the stomach: the lesser curvature part directly invaded into the lateral segment of the liver | 20cm×20cm×10cm | Abdominal fullness for 1–2 months and tenesmus for 2 weeks | Biphasic | EMA, cytokeratin, bcl-2 | Left lateral segmentectomy of the liver, subtotal gastrectomy with Billroth's II anastomosis and extensive resection of the left diaphragm tumor | Recurrence of multiple nodular lesions over the peritoneal area, perirectal area, left lateral region of the urinary bladder and mesenteric area after 2 months and died after 10 days |
Keratin, as an epithelial marker, is positive in about 90% of synovial sarcoma cases and is measured by immunohistochemistry. It's positivity along with histological correlation, aids diagnosis of synovial sarcoma.2
Epithelial Membrane Antigen (EMA) is another immunohistochemical marker, positive in majority of synovial sarcomas. Spindle cell components of the tumor, usually express Vimentin, and in up to 40% of cases, S-100 reactivity is identified.7 BCL-2 and CD99 markers often turn positive, while CD34 is mostly negative.6 In our reported case, Pan CK, Vimentin, EMA (figure C & D) and B-Catenin were positive but Inhibin, CDX2, CK7, CK20, ER, PR, Calretinin, CEA, CD10 and BCL2 were negative.
In over 90% of synovial sarcoma patients,5 a specific genetic abnormality, t (X;18) (p11.2;q11.2), and its resultant product, the SYT-SSX fusion protein is identifiable. The SSX gene is expressed on chromosome X, and is of three types: SSX1 in nearly 2/3rd of the cases, usually in biphasic sarcomas; SSX2 in a third of the cases, exclusively in monophasic type; and SSX4 which occurs rarely.8 These may only be detectable after surgery, with the use of pathology and immunohistochemical testing of the tumor. Use of cytogenetics and RT-PCR-based testing has shown to have increased sensitivity and specificity for diagnosing synovial sarcomas, through detection of the specific translocation.9 In our reported case, suggestion from the postoperative expert panel was to perform the complementary (X,18) translocation test. Study on remaining pelvic mass blocks was negative for SS18 (18q11) rearrangement but an extra copy of SS18 was reported in about 27% of tumor cells.
Management of this condition is challenging and therapy is controversial, given the rarity of the disease. Use of radiotherapy and chemotherapy after undergoing radical surgical resection has been described as a treatment.3 With regards to adjuvant chemotherapy for synovial sarcoma, Doxorubicin-Ifosfamide combination is the preferred first-line regimen. For treatment of advanced disease, Pazopanib (inhibitor of tyrosine kinase) and Trabectedin (an anticancer alkaloid agent)5 have been recommended recently.
No consensus has been made regarding the prognostic factors for the disease, though factors including biphasic tumor subtype, presence of SYT-SS1 fusion, location in extremities, small tumor size (<5cm), female gender, young age (<50 years) and negative surgical margins are reported in some studies to be associated with a more favorable prognosis.3
Information provided by intrinsic signal characteristics and superior soft tissue contrast, makes MRI the imaging modality of choice for synovial sarcoma diagnosis and initial staging. On T2-weighted images of MRI, the mixture of solid and cystic components, hemorrhagic areas, myxoid stroma, and fibrous tissue of a synovial sarcoma is revealed as a mosaic of combined low, intermediate, and high signal intensity. On CT scan, the tumor is featured as a mixed soft tissue with attenuation equal to or slightly lower than that of muscle. Necrotic or hemorrhagic change in the tumor present as heterogeneous areas. On T1-weighted MRI images, the tumor appears isointense or slightly hyperintense and on T2-weighted images it looks hyperintense to muscles. On CT and MRI imaging, presence of significant heterogeneity should rise the suspicion of synovial sarcoma.2
Up to 70% of patients with synovial sarcoma are reported to have local recurrence and distant metastasis. Metastases occur late in the course of the disease, mandating the need for long-term follow up. Lungs, liver and bone are the commonest sites of metastasis, in order of frequency. However, in contrast to most soft tissue malignancies, synovial sarcoma has small propensity for spread to lymph nodes. In Primary Ovarian Synovial Sarcoma, tumor recurrence has been observed up to 69 months, suggesting the need for long term surveillance, for early detection of local or distant tumor relapse.3 Primary intra-abdominal synovial sarcoma is a rare condition and is associated with high recurrence and fatality rates.2
ConclusionPrimary intra-abdominal synovial sarcoma is a rare neoplasm of aggressive nature with high mortality. The limbs, particularly the lower thigh and knee regions are the most common sites for occurrence of synovial sarcomas; occurrence of the disease at the other locations is infrequently reported. These neoplasms should be included in differential diagnosis of abdomino-pelvic masses in young and middle-aged patients.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Authors’ contributionsAll authors read and approved the final manuscript. All authors take responsibility for the integrity of the data and the accuracy of the data analysis.
FundingNone.
Availability of data and materialsThe data used in this study are available from the corresponding author on request.
Ethics approvalThis is a case study and does not contain any intervention with human participants or animals performed by any of the authors.
Informed consentThe patient informed consent has been obtained.
Consent for publicationBy submitting this document, the authors declare their consent for the final accepted version of the manuscript to be considered for publication.
Conflict of interestsThe authors declare that they have no competing interests.
