




La artrogriposis es un síndrome complejo, que responde etiológicamente a numerosas causas congénitas y adquiridas. Presentamos un caso familiar de artrogriposis. La necropsia aportó el hallazgo de inclusiones rojo-púrpura en el sarcoplasma del tejido muscular, sugestiva de miopatía por nemalinas.
Arthrogryposis is a complex syndrome, the aetiology of which can be traced to numerous congenital and acquired causes. We present a case of familiar arthrogryposis. Necropsy revealed red-purple rod-like structures in the sarcoplasm of the muscle tissue, suggestive of nemaline myopathy.
La artrogriposis no es un síndrome, si no, más bien un signo clínico complejo caracterizado por la presencia de contracturas y rigidez de las articulaciones, presente en más de 300 enfermedades diferentes. No es un diagnóstico médico sino la descripción de una limitación de movimiento de 2 o más articulaciones en diferentes zonas del cuerpo. Descrito por primera vez por Adolph Wilhelm Otto en 1841.
La miopatía nemalínica se caracteriza por una debilidad muscular, hipotonía y ausencia o disminución de reflejos tendinosos, su prevalencia es de 1/50.000 recién nacidos vivos (RNV) y su diagnóstico se realiza mediante biopsia muscular. Tiene un patrón de herencia autosómico dominante o recesivo.
Caso clínicoGestante de 37 años. G3C1P1 con antecedente de feto muerto intraútero a las 30 semanas por malformación fetal compatible con artrogriposis. En tratamiento con anticoagulantes a dosis profilácticas por presentar una mutación heterocigota en el gen de la protrombina.
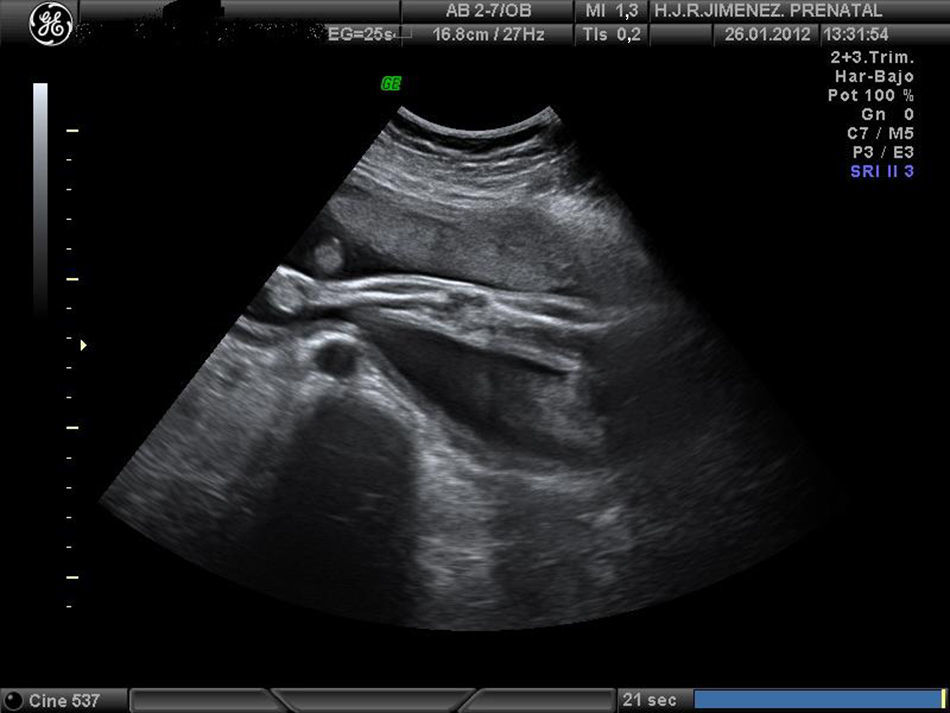
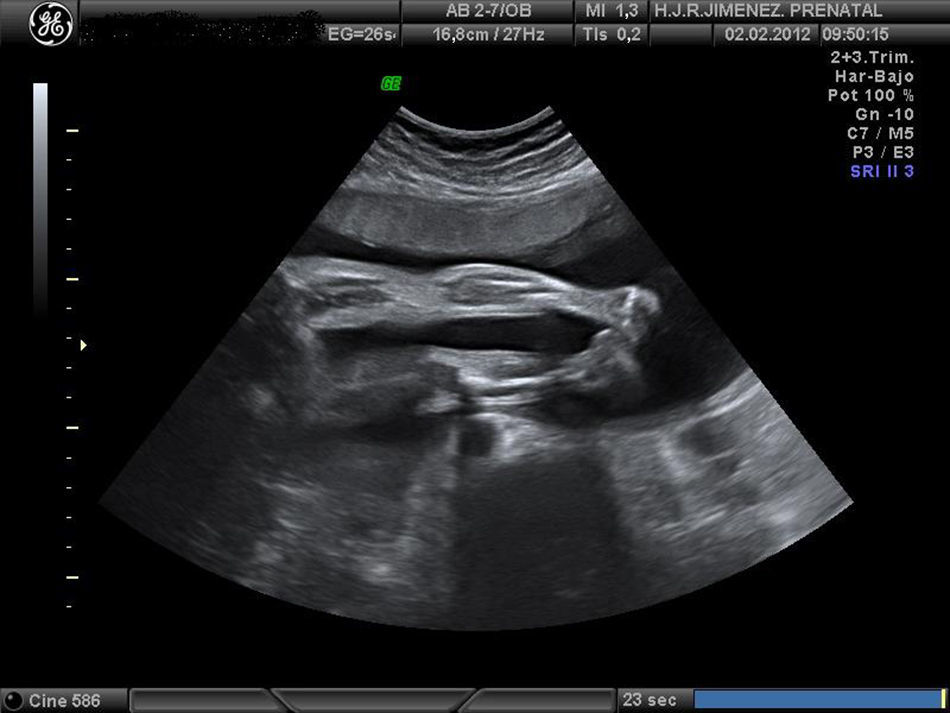
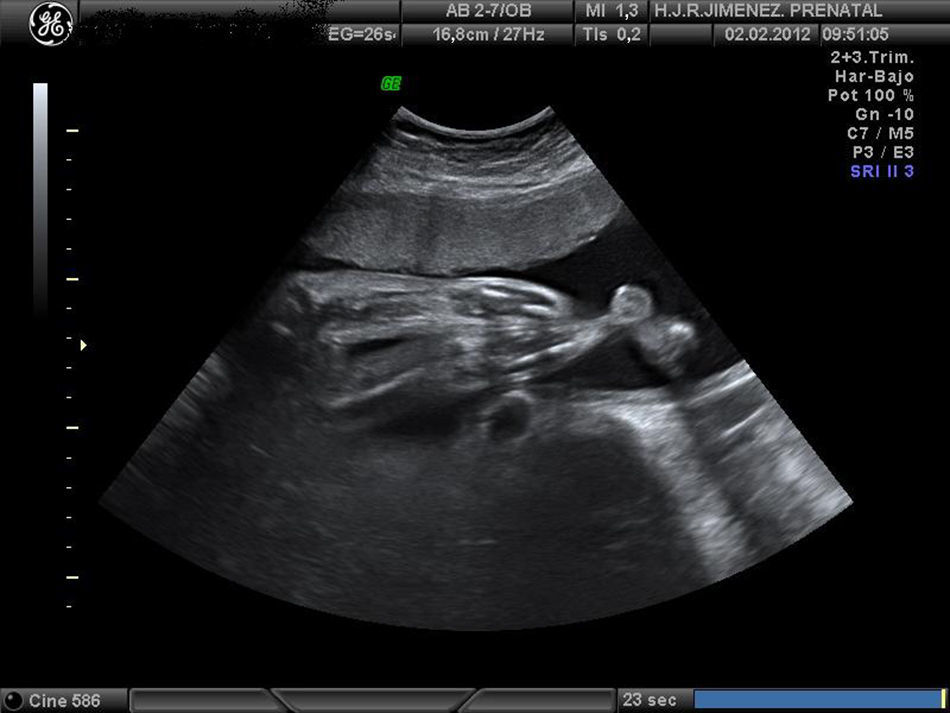
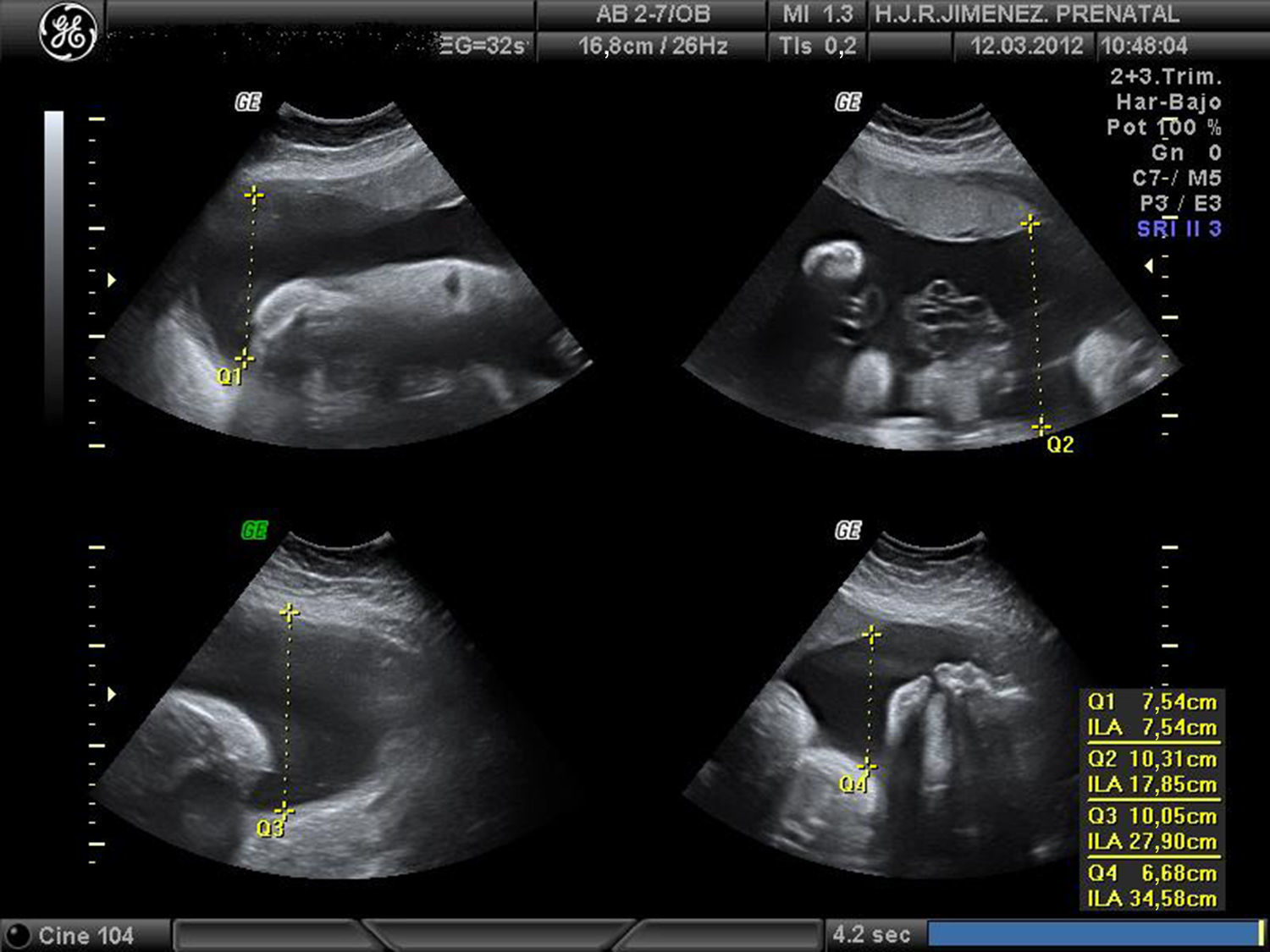
Controlada ecográficamente desde la 12+2 semanas, sin realizar cribado del primer trimestre por rechazo de la paciente. En la ecografía morfológica de la semana 20, se describe una posición mantenida en toda la exploración de las extremidades. A la 25+4 semanas se observa en la ecografía, feto con miembros inferiores en extensión y miembros superiores en flexión, y mantenidas durante la exploración (figs. 1-4). Se realiza amniocentesis diagnóstica con estudio de cariotipo normal, negativo para la detección del gen SMN1, para el despistaje de la atrofia muscular espinal. El estudio microarrays ampliado de enfermedades de depósito determinó el resultado negativo para gangliosidosis GM1, enfermedad de Gaucher, enfermedad de Pompe, enfermedad de I-CELL y mucopolisacaridosis tipo VII.




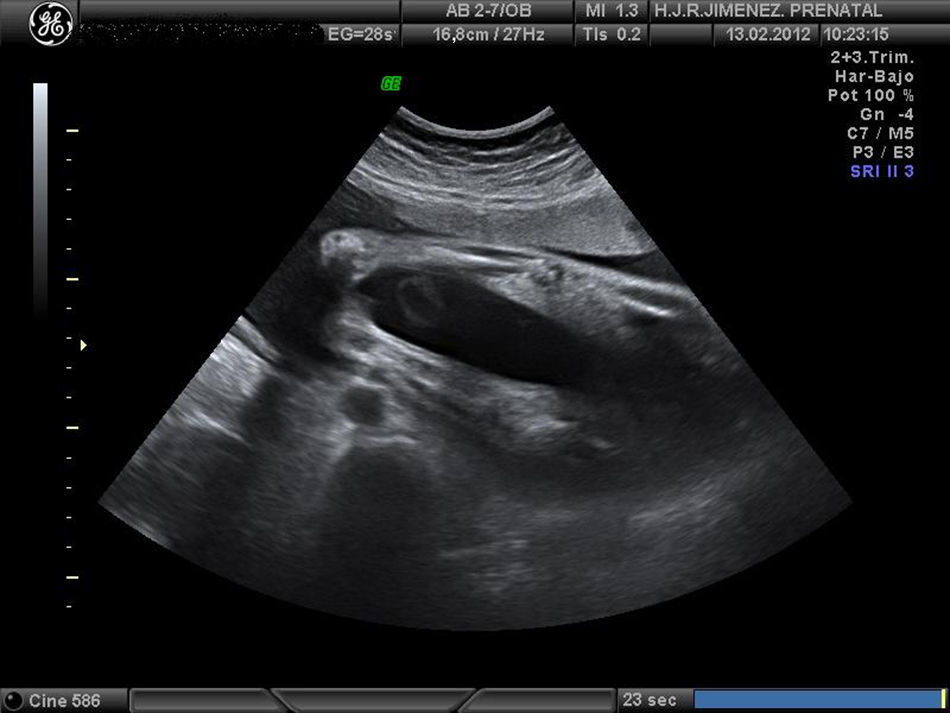
Alrededor de las 30 semanas se observa un polihidramnios (fig. 5) severo con movimientos fetales escasos y un ligero edema subcutáneo. Posteriormente a la semana 33, la paciente ingresa con sospecha de amenaza de parto pretérmino con una cervicometría de 20mm, funnel positivo y dinámica irregular. Se inicia tocolisis intravenosa, maduración pulmonar fetal y amniodrenaje terapéutico.

A las 33+5 semanas, finaliza la gestación con un parto de inicio espontáneo y final eutócico. Nace una mujer, con un Apgar 2/2 y sin respiración espontánea, con malformaciones aparentes. Se realiza RCP avanzada, sin éxito. Se extrae sangre y se realiza biopsia del músculo fetal para estudio.
El informe de anatomía patológica describe los siguientes hallazgos: «malformaciones a nivel macroscópico, con estigmas de artrogriposis». A nivel microscópico, el estudio muscular habla de una variabilidad en el tamaño de las fibras con distribución difusa, con una histoquímica de tricrómico positiva para inclusiones rojo-púrpura en sarcoplasma, sospechando de una miopatía de depósito de nemalinas, como causa de la artrogriposis.
El estudio genético de microarrays CGH, resultó normal. La determinación genética, que se realiza de rutina para la detección de dicha miopatía, resultó negativa para la presencia de los genes ACT1 y NEB. Actualmente no se realiza la determinación del resto de los marcadores genéticos asociados a la miopatía nemalínica, dada su baja incidencia.
DiscusiónLa artrogriposis está presente en más de 200 síndromes, y en algunos de estos síndromes las anomalías acompañantes —cardíacas, renales, pulmonares, etc.— son a veces tan graves que hacen incompatible la vida.
La incidencia actual de la artrogriposis se sitúa en 1:3.000-5.000RNV. No es un problema de la1 embriogénesis de la articulación, si no un defecto en su funcionalidad. Existen diferentes causas, unas de origen adquiridas y otras genéticas, como son la limitación del espacio intrauterino, los defectos del músculo esquelético, las miopatías, las neuropatías, las enfermedades maternas, el compromiso vascular; todas ellas cierran el cuadro etiológico de dicho síndrome.
Estas anomalías se caracterizan por su presencia2 ecográfica en el segundo trimestre, ya que es muy difícil visualizar en el3 primer trimestre contracturas articulares fijas, inmóviles, fundamentalmente de los miembros y, además, en posiciones bizarras. Suele ser este signo indirecto el que confirma finalmente al diagnóstico. Se trata, sobre todo, de contracturas en flexión y persistentes que crean deformaciones de las mismas. Estas se presentan de forma primaria o clásica, o integrando parte de otros síndromes o malformaciones. Aunque se han descrito casos con oligoamnios, el polihidramnios es muy frecuente y facilita el diagnóstico. También es habitual el hydrops fetalis4,5.
Centrándonos en la miopatía nemalínica como origen de la artrogriposis de nuestro caso clínico; el término «nemalínica» hace referencia a la presencia característica de bastoncillos o estructuras fibrilares (del griego nema, «hilo») en las fibras musculares6. La miopatía nemalínica es clínicamente heterogénea. Puede producirse una forma neonatal grave, que se caracteriza por hipotonía y dificultades para la alimentación que causan la muerte prematura. La biopsia de músculo indica la presencia de cúmulos de pequeños cilindros (cuerpos de nemalina), que aparecen preferentemente (aunque no de modo exclusivo) en las fibras musculares de tipo i. A veces, los cilindros también se observan en los mionúcleos. El músculo, por lo común, tiene predominio de fibras musculares de tipo i.
Se ha vinculado más de 5 genes con la miopatía por nemalina. Todos codifican proteínas asociadas a filamentos finos, lo cual sugiere la alteración del ensamblado o interrelación de estas estructuras como mecanismo fundamental. Las mutaciones del gen de la nebulina (NEB) explican casi todos los casos, incluyendo las formas graves neonatal y de la niñez temprana que se heredan por un mecanismo autosómico recesivo. Los casos en la fase neonatal e infantil que se heredan por mecanismos predominantemente autosómicos dominantes son causados por mutaciones del gen de la alfa actinina de músculo estriado (ACTA1)7. Formas menos graves de la enfermedad con mecanismo de herencia autosómico dominante se han identificado mutaciones en los genes de tropomiosina alfa lenta (TPM3) y tropomiosina beta (TPM2) que comprenden menos del 3% de los casos. Las mutaciones del gen de troponina T muscular (TNNT1) al parecer se circunscriben a la población amish en Estados Unidos. No se cuenta con tratamiento específico alguno8–10.
Se trata de una caso de asociación familiar de artrogriposis, con asociación familiar a la miopatía nemalínica. Posiblemente se trate de una alteración hereditaria y no una aparición esporádica pese al resultado negativo de los principales marcadores genéticos, dada la baja incidencia de dicha miopatía grave, y su aparición iterativa en la descendencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
