






El síndrome hemolysis (H), elevated liver enzimes (EL) y low platelets count (LP) (HELLP) es una microangiopatía trombótica propia del embarazo. Puede haber síndromes de HELLP incompletos. En éstos se pueden observar sólo algunos parámetros del síndrome: EL (enzimas hepáticas elevadas), ELLP (enzimas hepáticas elevadas y trombopenia) y LP (trombopenia aislada). El síndrome de ELLP supone un diagnóstico diferencial difícil en el que hay que descartar las principales causas de trombopenia en el embarazo y otras patologías. En la práctica clínica actual su manejo es similar al síndrome de HELLP completo. Sin embargo, la morbilidad materno-fetal es menor en el síndrome de ELLP. Esto podría sugerir la necesidad de valorar un protocolo de actuación distinto para estas dos variantes de una misma enfermedad.
HELLP syndrome (Hemolysis, Elevated Liver enzymes, and Low Platelet count) is a thrombotic microangiopathy of pregnancy. This syndrome may be incomplete, with manifestations of only a few of its parameters: EL (elevated liver enzymes), ELLP (elevated liver enzymes and thrombocytopenia) and LP (thrombocytopenia alone). ELLP syndrome is a difficult differential diagnosis in which the main causes of thrombocytopenia in pregnancy and other diseases must be excluded. In current clinical practice, the management of ELLP is similar to that of complete HELLP syndrome. However, maternal and fetal morbidity is lower in ELLP syndrome, which may suggest the need to evaluate different protocols for these two variants of the same disease.
El síndrome de ELLP es una microangiopatía trombótica específica de la gestación cuyo diagnóstico diferencial puede ser laborioso. Aquí describimos un caso y se revisa la bibliografía en búsqueda de la línea correcta para alcanzar dicho diagnóstico y el posterior manejo de esta patología.
Caso clínicoSe trata de una gestante nulípara de 29 años sin antecedentes de interés que fue remitida a las 30 semanas de gestación (sg) a las Consultas de Alto Riesgo Obstétrico de nuestro servicio con la sospecha diagnóstica de trombopenia gestacional. La paciente estaba asintomática, con cifras de tensión arterial (TA) normales y presentaba un recuento plaquetario de 97.000 plaquetas/mm3 siendo los previos normales. Se inicia un estudio de la trombopenia en colaboración con el Servicio de Hematología. En el siguiente control se objetiva una disminución de las plaquetas hasta 58.000 plaquetas/mm3 comprobada mediante recuento diferencial con EDTA y citrato, anticuerpos antiplaquetarios negativos, frotis de sangre periférica normal y proteinuria negativa. Dada la bajada de plaquetas y descartadas otras causas de trombopenia asociada a embarazo, se reorienta el diagnóstico hacia la púrpura trombocitopénica inmune (PTI). A las 37 sg la paciente acude a consulta presentando una medición de TA aislada de 180/80 mmHg y descenso de plaquetas hasta 45.000/mm3, por lo que se inicia tratamiento con inmunoglobulinas. Su respuesta no es óptima, por lo que se comienza pauta de corticoides. Una semana más tarde, la paciente vuelve a acudir a consulta refiriendo dolor epigástrico desde hace dos días. Sus controles de TA están en los límites de la normalidad. Sin embargo, en sus valores analíticos se observa un aumento de ácido úrico hasta 7,7 mg/dl, un aumento de transaminasas con GOT de 283 U/I y GPT de 428 U/I y un incremento de LDH hasta 713 U/I con cifras de hemoglobina, haptoglobina y bilirrubina total normales y ausencia de esquistocitos en sangre periférica. Las cifras de coagulación y fibrinógeno también son normales. Ante estos hallazgos se decide ingresar a la paciente y la realización de cesárea urgente con anestesia general por síndrome HELLP incompleto y Bishop desfavorable. Durante la cesárea y el postoperatorio no ocurre sangrado excesivo. Se obtuvo un recién nacido mujer de 2.390 g con Apgar 9-10 y trombopenia neonatal no inferior a 95.000 plaquetas/mm3.
En el control analítico materno poscesárea, el recuento plaquetario es de 14.000 plaquetas/mm3, por lo que se decide transfundir plaquetas y reiniciar tratamiento con dexametasona. Durante el puerperio se produce la normalización de los parámetros plaquetarios y de los niveles de enzimas hepáticas, por lo que se inicia pauta descendente de corticoides. Actualmente la paciente permanece asintomática, sin tratamiento y con controles ambulatorios analíticos normales.
DiscusiónEn 1982, Louis Weinstein introdujo estas siglas (H, EL y LP) para definir a un grupo de pacientes con o sin preeclampsia/eclampsia con estos hallazgos1. En 1991, Redman incluyó una modificación a estas siglas para aquellas pacientes en las cuales no se podía reconocer la anemia hemolítica y lo denominó ELLP2.
El síndrome HELLP se observa entre el 0,5 y el 0,9% de todas las gestaciones. La mortalidad materna asociada con HELLP se aproxima al 1-24%, y la perinatal al 40%, según diferentes autores. Audibert et al compararon las complicaciones de gestantes con ELLP y HELLP, las primeras tuvieron menor morbilidad (eclampsia 8 vs. 21%; lesiones cerebrales isquémicas 6 vs. 21%)3.
Ante la primera sospecha de trombopenia gestacional iniciamos el diagnóstico diferencial de trombopenia y embarazo. La trombopenia se define como un recuento plaquetario menor de 150.000 plaquetas/mm3 en dos determinaciones distintas. Las causas más comunes de trombopenia durante el embarazo son la trombopenia gestacional, el síndrome de HELLP, enfermedades autoinmunes e infecciones.
La trombopenia gestacional es la causa más común de trombopenia durante el embarazo y se presenta en el 5-8% de las mujeres embarazadas4. La disminución de las plaquetas se debe a hemodilución o aumento de su destrucción por la placenta. No suelen bajar de las 70.000 plaquetas/mm3, que vuelven a la normalidad en el posparto y puede reproducirse en siguientes gestaciones. Son asintomáticas. Se suele presentar al final del segundo y tercer trimestre del embarazo. Los anticuerpos antiplaquetarios no diferencian la trombopenia gestacional de la PTI, ya que en ambos cuadros los anticuerpos IgG antiplaquetarios pueden estar elevados y en el 25% de las PTI los anticuerpos antiplaquetarios específicos pueden estar ausentes. En nuestro caso la trombopenia gestacional fue la primera sospecha diagnóstica, pero se reorientó hacia la PTI por el descenso acusado de plaquetas hasta 45.000 plaquetas/mm3.
La PTI se caracteriza por una destrucción de plaquetas por anticuerpos IgG. Es la causa más frecuente de trombopenia clínicamente significativa durante el primer trimestre. Se caracteriza por recuentos plaquetarios significativamente inferiores a 70.000 plaquetas/mm3, ausencia de esplenomegalia y exclusión de enfermedades sistémicas o drogas que causen trombopenia. El neonato puede desarrollar PTI por transferencia transplacentaria de anticuerpos IgG maternos con el consiguiente riesgo aumentado de hemorragia intraventricular. En nuestro caso, tras descartar la trombopenia gestacional se inició el manejo de la paciente según el protocolo de PTI comenzando el tratamiento con corticoides e inmunoglobulinas. Sin embargo, ante la aparición de epigastralgia y la elevación de enzimas hepáticas asociado a la trombopenia sin signos de hemólisis, se etiqueta finalmente la patología de nuestra paciente como síndrome HELLP incompleto.
En la actualidad coexisten dos sistemas de clasificación para el diagnóstico de síndrome HELLP5,6 (tablas 1 y 2). De acuerdo con la clasificación de Tennesse realizada por Sibai5, puede haber síndromes de HELLP incompletos. En éstos se pueden observar sólo algunos parámetros del síndrome: EL, ELLP y LP.
Clasificación del síndrome HELLP del Centro Médico de la Universidad de Mississippi
| Trombocitopenia | Hemólisis+disfunción hepática |
| Clase 1:≤500.000 plaquetas/mm3 | LDH≥600 U/I |
| Clase 2: > 50.000 plaquetas/mm3 ≤ 100.000 plaquetas/mm3 | AST y/o ALT≥40 U/I |
| Clase 3: > 100.000 plaquetas/mm3 ≤ 150.000 plaquetas/mm3 | |
| Deben estar presentes todos los parámetros para poder ser clasificados | |
Clasificación del síndrome HELLP de la Universidad de Tennessee
| Síndrome de HELLP completo | Síndrome de HELLP incompleto |
| Hemólisis | Sólo uno o dos de los criterios presentes |
| Esquistocitos, esferocitos o fragmentos de hematíes en frotis periférico | |
| LDH≥600 U/I | |
| Bilirrubina indirecta > 1,2 mg/dl | |
| Elevación de enzimas hepáticas | |
| LDH≥600 U/I | |
| AST y/o ALT≥72 U/I | |
| Trombocitopenia | |
| < 100.000 plaquetas/mm3 | |
Una de las claves reside en establecer los criterios diagnósticos de hemólisis para poder excluirla. Muchos autores han utilizado los niveles de LDH total como criterio diagnóstico de hemólisis. Sin embargo, hay cinco isoformas de LDH y solamente dos de ellas (LDH 1 y LDH 2) son liberadas de forma mayoritaria por la destrucción de hematíes. Así, en la mayoría de las mujeres con preeclampsia severa/eclampsia, la elevación de LDH total sea causada, probablemente, por el daño hepático. Por lo tanto, Sibai aboga por las alteraciones del frotis de sangre periférica (esquistocitos, esferocitos o fragmentos de hematíes), el descenso de haptoglobina y la elevación de bilirrubina indirecta para definir la hemólisis. Algunos autores proponen comprobar la hemólisis si los valores de haptoglobina descienden por debajo de 0,3 mg/dl7. La haptoglobina es una glucoproteína que transporta la hemoglobina hasta su sitio de degradación. Cuando hay una hemólisis anormal, la hemoglobina liberada se une a la haptoglobina disminuyendo su concentración.
La fisiopatología del HELLP es poco conocida. Se asocia a lesión endotelial con deposición de fibrina en los vasos sanguíneos, incremento del consumo plaquetario. Cuando la fibrina se deposita en los sinusoides hepáticos, los obstruye, produce daño hepatocelular con la consiguiente elevación de enzimas hepáticas, dolor epigástrico y a nivel de hipocondrio derecho.
La clínica de epigastralgia que presentó nuestra paciente se ajusta a la esperada en los HELLP completos: en el 90% de los casos los primeros síntomas consisten en dolor epigástrico o en hipocondrio derecho. La hipertensión y la proteinuria pueden ser leves e incluso ausentes (en el 20-30% de casos, respectivamente), lo que retrasa el diagnóstico7.
El síndrome de ELLP se asocia a un aumento de morbilidad materna (coagulación intravascular diseminada, desprendimiento prematuro de placenta normoinserta, etc.) y neonatal (distrés respiratorio, hemorragia cerebral, etc.), pero siempre inferior al síndrome HELLP3. En nuestro caso, el recién nacido presentó trombopenia neonatal, una complicación rara pero descrita en los síndromes de HELLP5,8.
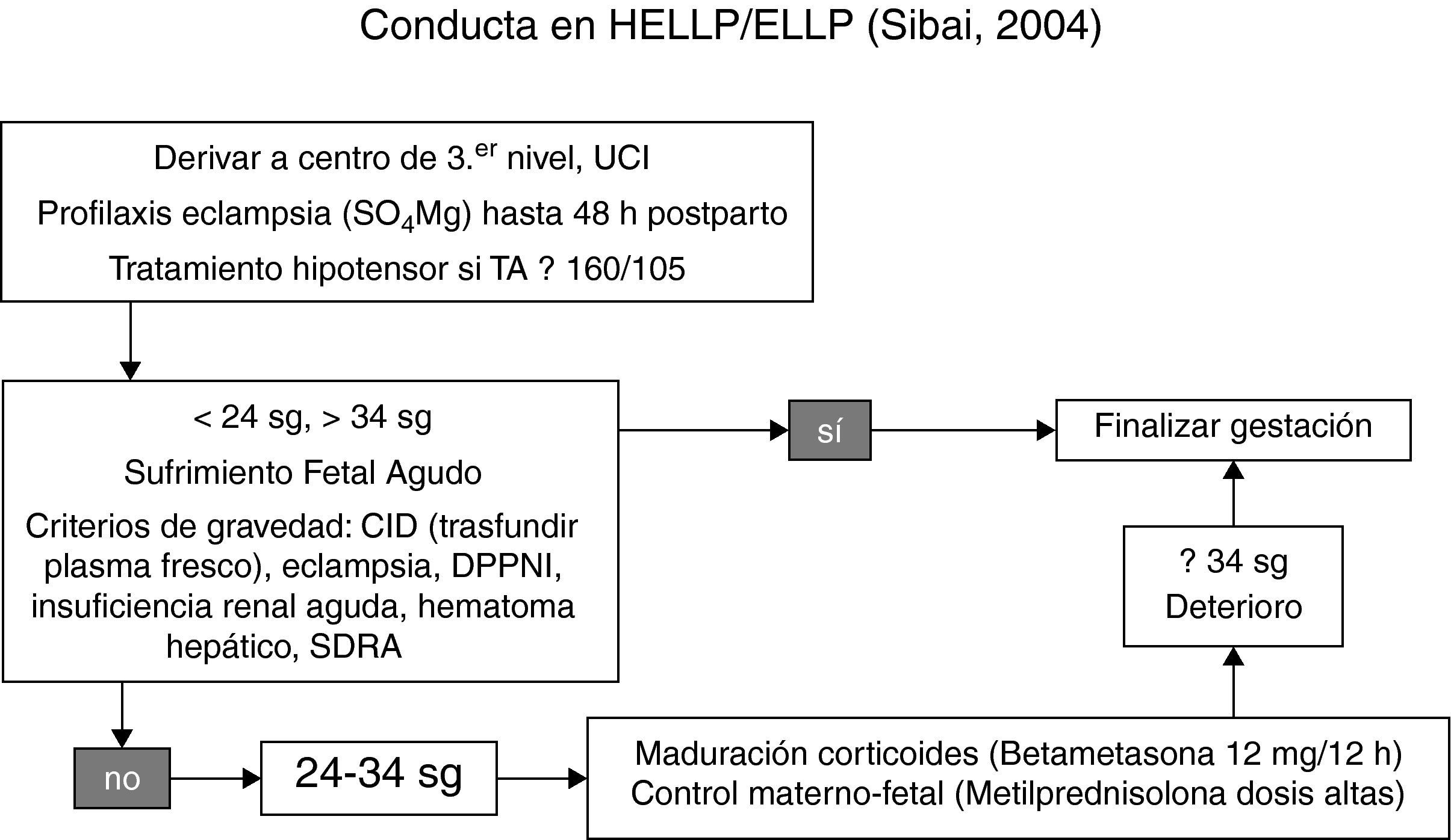
Las pacientes con síndrome de ELLP llevan a plantearse algunos interrogantes como: ¿cambia la conducta clínica si se trata de un ELLP con respecto a un HELLP? Lo que se observa en la práctica clínica actual es que la conducta es la misma9: el manejo de las pacientes según la edad gestacional y la severidad del cuadro clínico5 (fig. 1).
.")
Si una paciente presenta recuentos plaquetarios por debajo de 20.000 plaquetas/mm3 debe transfundirse concentrados de plaquetas. La administración de corticoides en el posparto se dirige a evitar un descenso plaquetario y la elevación de enzimas hepáticas.
ConclusionesEl síndrome de ELLP supone un diagnóstico diferencial difícil en el que hay que descartar las principales causas de trombopenia en el embarazo y otras patologías. En la práctica clínica actual su manejo es similar al síndrome de HELLP completo. Sin embargo, la morbilidad materno-fetal es menor en el síndrome de ELLP. Esto podría sugerir la necesidad de valorar un protocolo de actuación distinto para estas dos variantes de una misma enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
