





The aim of the present study was to examine the probable relationship between the accumulation of oxLDL and hepatic fibrogenesis in cholestatic rats.
INTRODUCTIONThere is growing evidence to support the current theories on how oxidative stress that results in lipid peroxidation is involved in the pathogenesis of cholestatic liver injury and fibrogenesis. One of the major and early lipid peroxidation products, OxLDL, is thought to play complex roles in various immuno-inflammatory mechanisms.
METHODSA prolonged (21-day) experimental bile duct ligation was performed on Wistar-albino rats. Biochemical analysis of blood, histopathologic evaluation of liver, measurement of the concentration of malondialdehyde (MDA) and superoxide-dismutase (SOD) in liver tissue homogenates, and immunofluorescent staining for oxLDL in liver tissue was conducted in bile-duct ligated (n = 8) and sham-operated rats (n = 8).
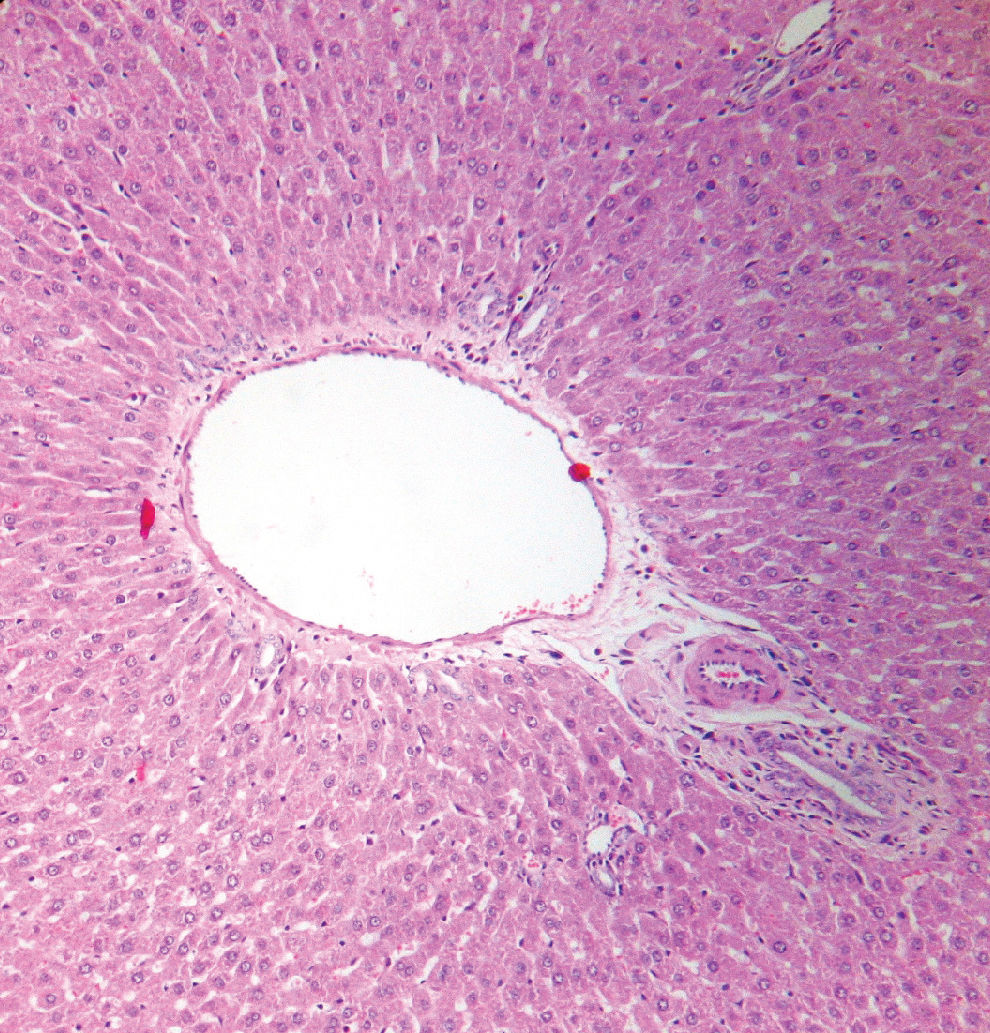
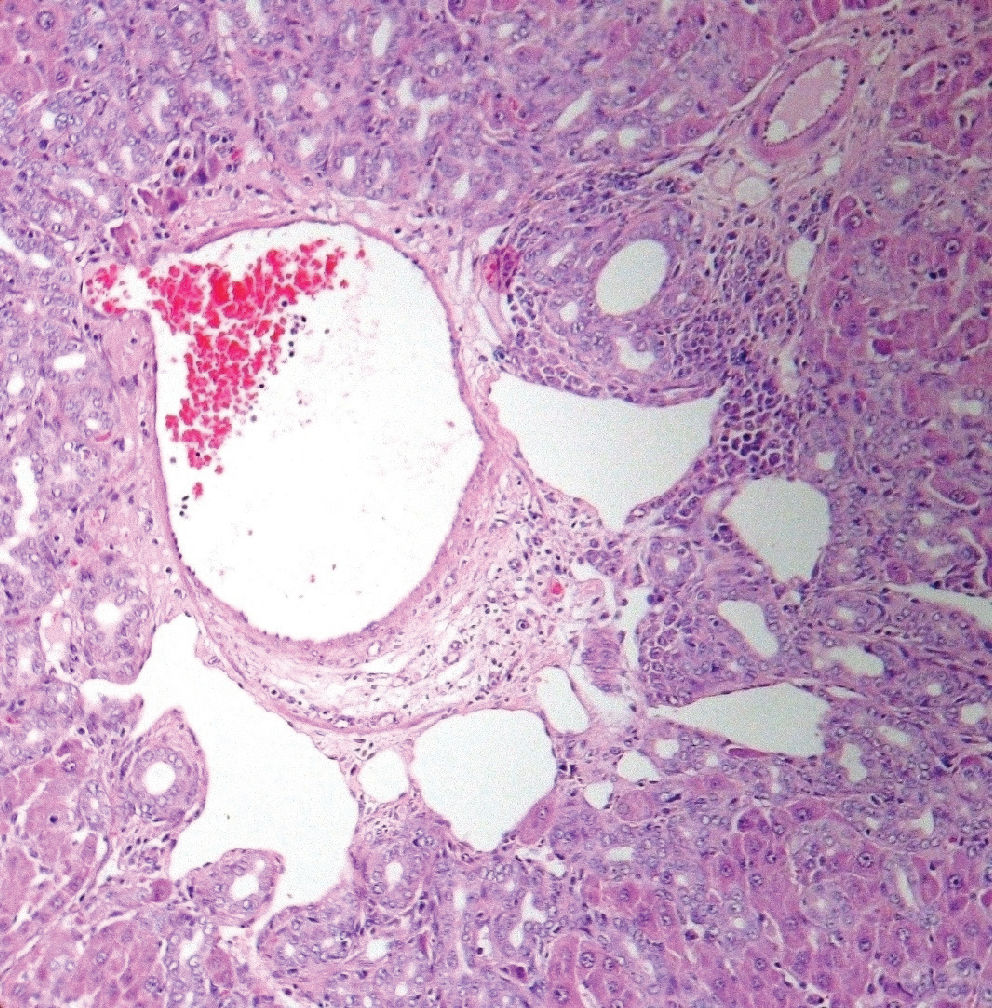
RESULTSSignificantly higher levels of MDA and lower concentrations of SOD were detected in jaundiced rats than in the sham-operated rats. Positive oxLDL staining was also observed in liver tissue sections of jaundiced rats. Histopathological examination demonstrated that neither fibrosis nor other indications of hepatocellular injury were found in the sham-operated group, while features of severe hepatocellular injury, particularly fibrosis, were found in jaundiced rats.
CONCLUSIONOur results support the finding that either oxLDLs are produced as an intermediate agent during exacerbated oxidative stress or they otherwise contribute to the various pathomechanisms underlying the process of liver fibrosis. Whatever the mechanism, it is clear that an association exists between elevated oxLDL levels and hepatocellular injury, particularly with fibrosis. Further studies are needed to evaluate the potential effects of oxLDLs on the progression of secondary biliary cirrhosis.
A prolonged obstruction of either the intra- or extra-hepatic biliary duct may damage hepatic tissue and lead to the development of fibrosis and cirrhosis.1 Liver repair and fibrogenesis both resemble the wound healing process. Cirrhosis is an advanced stage of fibrosis that is characterized by the formation of regenerative nodules in the liver parenchyma separated by fibrotic septa. The generation of cirrhosis involves three major processes: cell death, aberrant extracellular matrix deposition (fibrosis), and vascular reorganization.2 Cholestasis is associated with, and probably best defined as, the impairment of bile formation. This impairment may be accompanied by or caused by the failure to excrete bile acids into the hepatocyte canaliculus, which disrupts the circulation of enterohepatic bile acid and elevates concentrations of hepatocellular and serum bile acid.3
The impairment in hepatic regeneration in cholestatic livers is a well-known phenomenon that may be caused by altered hepatic hemodynamics (decreased portal venous flow), attenuated production of mediators that are associated with liver proliferation, increased rate of apoptosis, and blocked enterohepatic circulation. These processes are all induced by biliary obstruction,4 which reduces the portal venous flow and the production of liver growth-associated factors, increases hepatocyte apoptosis, and impairs enterohepatic circulation. All of these factors attenuate the hepatic regenerative capacity. The intrahepatic accumulation of toxic bile salts results in hepatocyte apoptosis via a Fas-dependent mechanism.5 Many toxic bile acids trigger oligomerization of the Fas death receptor, which, in turn, activates the classic Fas death receptor cascade.6 In addition, it has been shown that an increased expression of Toll-like receptor 2 and the Fas ligand in intrahepatic natural killer T cells after biliary obstruction is associated with hepatocyte apoptosis.7 These results indicate that biliary obstruction activates hepatocyte apoptosis. Bile acids may directly affect mitochondria, causing mitochondrial dysfunction and activating the mitochondrial pathway of cell death.8 This information suggests that bile acids induce apoptosis in vitro.
Fibrosis is characteristic of chronic liver disease, irrespective of the underlying etiology. Hepatic stellate cells (HSCs) trigger pathogenesis by a liver damage-dependent activation.9 Activated HSCs transdifferentiate into myofibroblasts (MFBs), which exhibit a synthesis profile that predisposes them to increased extracellular matrix (ECM) deposition. This process allows them to proliferate upon expression of mitogenic cytokines and their receptors, alters their morphology, and increases contractility by triggering - smooth muscle actin (SMA) fiber formation. This then leads to the constriction of sinusoidal blood flow and increases the synthesis and release of fibrillar collagens.10
Myofibroblasts do not normally exist in the liver. They are produced by the activation of HSCs and portal fibroblasts. Myofibroblasts (MFBs) derived from small portal vessels proliferate around biliary tracts in cholestasis-induced liver fibrosis to initiate collagen deposition.11
Fibrosis develops with different spatial patterns according to the different prevalent mechanisms that are associated with the diverse causes of parenchymal damage. Indeed, fibrosis that is caused by chronic viral infection is initially concentrated within and around the portal tract, whereas fibrosis that occurs as a secondary effect of exposure to toxic/metabolic damage (as in bile duct obstruction) is located mainly in the centrolobular areas. Additionally, it is becoming increasingly evident that different cell types are involved in the deposition of the fibrillar extracellular matrix during active hepatic fibrogenesis. Hepatic stellate cells are mainly involved when hepatocellular damage is limited or concentrated within the liver lobule, whereas portal MFBs and fibroblasts contribute the most when the damage is located in the proximity of the portal tracts. In the later stages of evolution (septal fibrosis), it is likely that all extracellular matrix-producing cells will contribute to fibrogenesis.12
The reduction of expression and function of transport systems is another crucial process that plays a key role in the pathogenesis of cholestasis and may cause or maintain cholestatic injury.13,14 Transport defects may be caused by hereditary genetic defects or acquired as a result of cholestatic injury, such as during inflammation, drug use, or biliary obstruction. It is also important to keep in mind that not all of the encountered changes in transporter expression are procholestatic and negative. While some of these alterations contribute to cholestasis, other changes might act as compensatory anticholestatic defense mechanisms, providing alternative excretory routes. Reducing basolateral bile acid uptake while simultaneously increasing basolateral bile acid excretion might be considered a major hepatic defense mechanism counteracting bile acid accumulation within hepatocytes.15
With regard to the pathogenesis of cholestatic liver injury, the oxidative stress that is associated with increased lipid peroxidation is considered to be crucial for hepatotoxicity.16,17 Lipid peroxidation, protein oxidation, and the thiol redox state of the cell are important indicators of oxidative stress.18 Bile duct ligation causes changes in the equilibrium between the antioxidant and prooxidant activities; it favors the latter, as it increases production of free radicals and causes the reduction in free radical scavengers, such as catalase, superoxide dismutase, and glutathione peroxidase 19
The accumulation of hydrophobic bile acids impairs the mitochondrial electron transport chain and enhances the production of reactive oxygen species (ROS), which eventually disturb cellular energy homeostasis.20,21 Lipid peroxidation causes the depletion of mitochondrial respiratory chain elements, leading to a vicious cycle that results in more ROS generation.22
Lipids are known to be one of the major and consistent targets of oxidative stress.1 Studies on animals and humans have shown that there is a close relationship between lipid peroxidation and cholestatic liver injury.21,23 The role of lipid peroxidation products in the pathogenesis of excessive fibrogenesis in cholestasis has been, so far, the most investigated aspect of oxidative stress-induced damage. One of the major and early lipid peroxidation products to be identified was oxidized low-density-lipoprotein (oxLDL), which was first identified in large vessel walls when antioxidative protective pathways had failed; it was later popularized as a proatherogenic agent. Since then, oxLDL has been considered to be very important either in the initiation or perpetuation of atherosclerotic processes.24 Following the identification of specific membrane receptors for oxLDL on Kupffer cells, HSCs, macrophages, and liver endothelial cells, we previously showed that oxLDL accumulates in the liver of bile duct-ligated BALB/c mice.25,26 Here, we used a prolonged (21-day) experimental bile duct ligation to examine the probable relationship between oxLDL accumulation and hepatic fibrogenesis in rat liver.
MATERIALS AND METHODSAnimalsSixteen male albino Wistar rats, weighing 200–230 g, were used in the study. They were housed in stainless steel cages, under controlled temperature (23°C) and humidity with 12-hour dark/light cycles. They were maintained on a standard laboratory diet with tap water ad libitum throughout the experiment, except for an overnight fast before surgery. The experiments were carried out according to the guidelines set forth by the Ethics Committee of Zonguldak Karaelmas University, Zonguldak, Turkey.
Experimental DesignAnimals were randomly divided into two groups of 8 rats each. Group I was designated as the prolonged obstructive jaundice group (OJ), while group II was established as the sham-operated group (SO). All surgical procedures were performed under strict sterile conditions, using intraperitoneal ketamine-HCl (Ketalar; Parke Davis, Morris Plains, NJ, USA) as the anesthetic agent (50 mg/kg bodyweight) and a standard upper midline incision. In group I, a bile duct ligation was carried out according to the method of Lee.27 In group II, the sham operation was performed by placing a silk ligature around the common bile duct and then removing it. The abdominal incision was closed in two layers with chromic 4–0 cat gut and 4–0 silk. The rats were allowed food and water ad libitum postoperatively. On the 21st day after the operation, blood samples of all animals were collected using the cardiac puncture method. Blood was centrifuged at 1,800 g for 15 min at 4°C to obtain plasma for biochemical analysis. After blood collection, the rats were killed and the livers of the animals were removed immediately for histopathological, immunofluorescent, and biochemical examination.
Biochemical analysis of bloodThe serum bilirubin level was determined with Cobas Bio (Hoffman La Roche, Basel, Switzerland) using the bilirubin test (Hoffman La Roche). The serum activity of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), albumin, and gamma glutamine transpeptidase (GGT) was estimated using commercially available kits (Boehringer, Mannheim, Germany). Total protein concentration was measured according to the method described by Lowry et al, using serum bovine albumin as a standard.28
Malondialdehyde (MDA) and superoxide dismutase (SOD) measurement in liver tissue homogenateThe liver MDA level was assessed according to the method described by Ohkawa and Ohishi.29 In brief, the liver was homogenized with cold 1.15% KCl to make a 10% homogenate. A solution of 0.2 ml of 8.1% SDS and 1.5 ml of 20% acetic acid was adjusted to pH 3.5 with NaOH, and 1.5 ml of 0.8% aqueous solution of thiobarbituric acid was added to 0.2 ml of 10% tissue homogenates. The volume of the mixture was adjusted to 4.0 ml with distilled water, and then the mixture was heated in an oil bath at 95°C for 60 min. After cooling with tap water, 1.0 ml of distilled water and 5.0 ml of a mixture of n-butanol and pyridine (15:1, v/v) were added, and the solution was shaken vigorously. After centrifugation at 4,000 rpm for 10 min, the organic layer was removed and its absorbance was measured at 532 nm on a Shimadzu UV 1601 spectrophotometer. As a standard, 1.1.3.3-tetraetoxypropane was used. The MDA concentration was calculated per gram of tissue (nmol/gr tissue).
Hepatic SOD level was determined using the method of Sun et al.30 The assay for SOD activity involves inhibiting the reduction of nitroblue tetrazolium with xanthine and xanthine oxidase, which was used as a superoxide generator. Liver tissues were weighed, and 1.15% KCL was added to make a 10% homogenate. To 0.5 ml of the supernatant, 2.45 ml SOD measurement reactive solution (40 ml 0.3 mmol/L xanthine, 20 ml 0.6 mmol/L EDTA, 20 ml 150 μmol/L nitroblue tetrazolium, 12 ml 400 mmol/L Na2CO3, and 6 ml 1gr/L bovine serum albumin) was added. At 25°C, xanthine oxidase reagents were added in a volume of 0.05 ml per tube at 30 sec intervals and incubated for 20 min. The xanthine oxidase reagent was freshly prepared with ice-cold 2 mol/L NH4SO4; the final concentration of xanthine oxidase was 167 U/L. Reactions were then terminated by adding 1 ml of 0.8 mmol/L CuCl2 reagent into each tube at 30 sec intervals. The absorbance of each sample was measured at 560 nm on a Shimadzu UV 1601 spectrophotometer. The percent inhibition was calculated according to the formula:

One unit of SOD is defined as the amount of protein that inhibits the rate of NBT reduction by 50%. SOD activity was calculated per mg of tissue protein (U/mg protein).
Immunofluorescence staining of the rat liver tissueThe presence of oxLDL in the liver tissue sections of the jaundiced and the sham-operated rats was evaluated using a special method of immunofluorescence staining. The left lobe of each rat liver was obtained and stored at −85°C in a deep freezer. The slides were prepared from frozen liver biopsy sections, which were cut to a thickness of 7 μm. Slides were further divided into two pieces: one was used for the test, and the other was used as a negative control. Primary antibody in 30 μl (human polyclonal anti-oxLDL immunoglobulin (Ig)G, IMMCO Diagnostics, New York, NY, USA) was added only to the test slides, wheras phosphate buffered solution (PBS) in the same volume was added to the control slides. After incubating for 30 min in a humid chamber at room temperature, both the control and test slides were washed with PBS, and a fluorescein isothiocyanate labeled antihuman IgG (30 μl) was administered as a conjugate substance. After another 30 min at room temperature, the slides were washed with the standard PBS solution. After drying, the slides were covered with a mounting medium and examined under a fluorescence microscope (Leica DMRX, Wetzlar, Germany).
Histopathological evaluationFor histological examination, tissue samples from the liver were obtained from all animals. The liver samples were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. A single pathologist, blind to the study, assessed all biopsies. Although we acknowledge that there are differences in the pathogenesis of cholestasis cases caused by obstruction and hepatocellular lesions caused by chronic hepatitis, we have herein evaluated a scoring system following the same categorization as described by Ishak et al. in order to provide semi-quantitative scores to assess the degree of inflammatory activity and fibrosis.31 Liver damage was categorized as mild, moderate, or severe (cirrhosis). Inflammatory scores between 1–3 and fibrosis scores between 1–2 were classified as mild; inflammatory scores between 4–18 and fibrosis scores between 3–5 were classified as moderate liver damage. Cirrhosis was considered to be a fibrosis score of 6.
Statistical analysisData are expressed as mean ± SD. Comparisons among the groups were performed using the Mann-Whitney U test. Differences were considered significant when p < 0.05.
RESULTSNo deaths were observed in groups I or II. All animals with bile duct ligation were obviously jaundiced by 5–6 days after the operation. The jaundice was confirmed by assessing serum bilirubin concentration on the 21st day after bile duct ligation.
The mean values of the serum AST, ALT, ALP, GGT, total protein, bilirubin, and albumin concentrations measured in the blood samples are presented in Table 1. Prolonged obstructive jaundice significantly increased the serum AST, ALT, ALP, GGT, and bilirubin concentrations as expected. While there were no statistically significant differences between groups in serum total protein concentration, serum albumin concentrations were significantly decreased in prolonged obstructive jaundiced animals.
Mean values of the serum biochemical analysis
| Group | Albumin (g/dl) | ALP (U/L) | ALT (U/L) | AST (U/L) | Bilirubin (mg/dl) | TP (g/dl) | GGT (U/L) |
|---|---|---|---|---|---|---|---|
| 1 (n=8) | 2,84±0,41* | 254,73±36,23* | 107,75±34,89* | 452,38±148,15* | 10,35±3,82* | 7,48±0,60 | 28,9±27,77* |
| 2 (n=8) | 3,74±0.53 | 187,90±43,36 | 64,38±15,77 | 199,13±58,20 | 0,47±0,17 | 7,39±0,25 | 0,95±0,76 |
p < 0.05 using Mann-Whitney U test. Values are mean ± SD; Group 1: prolonged obstructive jaundiced group. Group 2: sham-operated rats. Serum ALP, ALT, AST, GGT, and bilirubin levels were determined to be significantly increased in group I, whereas albumin level was found to be significantly decreased.
The mean MDA and SOD activities of the livers in each group are shown in Table 2. While the mean liver content of SOD in group I was significantly lower than that of group II, prolonged bile-duct ligation resulted in a significant increase in the liver MDA content as compared to sham-operated rats (Table 2).
Mean liver MDA and SOD concentrations in the prolonged obstructive jaundiced and sham-operated rats
| Group | MDA (nmol/gr liver) | SOD (U/mg protein) |
|---|---|---|
| 1 (n=8) | 35.07±3.73* | 1.62±1.36* |
| 2 (n=8) | 20.86±4.69 | 7.86±1.42 |
p < 0.01 using Mann-Whitney U test. Values are mean ± SD; Group 1: prolonged obstructive jaundice group. Group 2: sham-operated rats. Twenty-one days of biliary obstruction causes a significant increase in MDA level, while hepatic SOD concentration was significantly lower in group 1 than in group 2.
A significant positive immunofluorescence staining for oxLDL was found in the liver tissue sections of the prolonged jaundiced group (group I; Figure 1), and we did not observe any positive immunofluorescence staining in sham-operated rats (group II; Figure 2).


Histopathological sections obtained from the liver tissues were examined under a light microscope. The evaluation of the above-mentioned criteria was performed by the same histopathologist. Neither fibrosis nor any of the other criteria for inflammation and hepatocellular injury were observed in the sham-operated group, whereas features of severe hepatic injury, namely fibrosis, were determined in the liver tissues of prolonged obstructive jaundiced rats (Figures 3–4). Table 3 displays the histological evaluation.


Histological activation index modified by Ishak et al.
| GRADE: Necroinflammatory score (0–18) | STAGE: Fibrosis score (0–6) | ||||
|---|---|---|---|---|---|
| Peri- portal/periseptal interface hepatitis | Confluent necrosis | Focal lytic necrosis, apopitosis, focal inflammation | Portal inflammation | Fibrosis | |
| OJ-1 | 3 | 1 | 1 | 2 | 4 |
| OJ-2 | 3 | 2 | 2 | 3 | 3 |
| OJ-3 | 3 | 3 | 2 | 3 | 3 |
| OJ-4 | 3 | 3 | 2 | 4 | 4 |
| OJ-5 | 3 | 2 | 3 | 3 | 3 |
| OJ-6 | 3 | 2 | 2 | 2 | 4 |
| 0J-7 | 3 | 2 | 2 | 3 | 4 |
| OJ-8 | 2 | 3 | 2 | 2 | 3 |
| SO-1 | 0 | 0 | 0 | 0 | 0 |
| SO-2 | 0 | 0 | 0 | 0 | 0 |
| SO-3 | 0 | 0 | 0 | 0 | 0 |
| SO-4 | 0 | 0 | 0 | 0 | 0 |
| SO-5 | 0 | 0 | 0 | 0 | 0 |
| SO-6 | 0 | 0 | 0 | 1 | 0 |
| SO-7 | 0 | 0 | 0 | 0 | 0 |
| SO-8 | 0 | 0 | 0 | 0 | 0 |
OJ = obstructive jaundiced rat; SO = sham-operated rat
Obstructive jaundice leads to hepatocellular injury and causes many systemic complications. Irrespective of the etiology, liver fibrosis is a common consequence of chronic liver injury.32 Liver fibrosis is defined as the abnormal accumulation of extracellular matrix in the liver. Its endpoint is cirrhosis, which is responsible for significant morbidity and mortality.
Recently, hereditary defects in the hepatobiliary transporter have been shown to cause cholestasis. Cholestasis may result either from a functional defect in bile formation at the level of the hepatocyte or from an impairment in the secretion of bile and/or obstruction of flow at the bile duct level.33
Hepatic uptake and biliary excretion of organic anions, such as bile acids and bilirubin, is mediated by hepatobiliary transport systems. Defects in transporter expression and function may cause or maintain cholestasis and jaundice. The recruitment of alternative export transporters in coordination with phase I and II detoxifying pathways provides alternative elimination pathways to counteract the accumulation of potentially toxic biliary constituents in cholestasis.32 The genes encoding for organic anion uptake (NTCP/SLC10A1, OATPs), canalicular export (BSEP, MRP2), and alternative basolateral export (MRP3, MRP4) transporters in the liver are regulated by a complex interacting network of hepatocyte nuclear factors (HNF1, 3 and 4) and nuclear receptors (e.g., FXR, PXR, CAR, RAR, LRH-1, SHP, and GR).34 Moreover, adaptive transporter changes, such as the induction of basolateral efflux pumps, may help to limit hepatic accumulation of potentially toxic biliary constituents in cholestasis by providing alternative escape routes.33 Bile acids, proinflammatory cytokines, hormones, and drugs mediate these causative and adaptive transporter changes at a transcriptional level by interacting with these nuclear factors and receptors.
Increased matrix formation (i.e., fibrogenesis) occurs in parallel to decreased matrix degradation (i.e., fibrolysis). A landmark finding was the identification of activated HSCs and MFBs as the major collagen-producing cells, which, upon activation by profibrogenic cytokines, acquire a profibrogenic phenotype and start to synthesize extracellular matrix that replaces parenchymal tissue.35 The progression of fibrosis results in the development of cirrhosis, chronic liver failure, portal hypertension, and hepatocellular carcinoma.31,36
As fibrosis develops in response to liver injury, HSC activation leads to the accumulation of a scar matrix. This process, in turn, contributes to the loss of hepatocyte microvilli and endothelial fenestrae, leading to the deterioration of hepatic function. Kupffer cell (macrophage) activation contributes to the activation of HSCs.37 In the cholestatic rodent, Kupffer cell activation and the infiltration of neutrophils contribute to tissue injury.38 Similarly, in our experimental data, neutrophil infiltration was evident in bile duct-ligated rats but not in sham-operated rats.
Activated HSCs, portal fibroblasts, and MFBs of bone marrow origin have been identified as the major collagen-producing cells in the injured liver. These cells are activated by fibrogenic cytokines, such as angiotensin II, TGF-1, and leptin. 39
Recent reports have highlighted the important role of bile in maintaining intestinal integrity and normal intestinal immunity.40 The impaired function of the intestinal barrier in patients with biliary obstruction is restored by bile replacement.41 Bile administration maintains the integrity and permeability of the intestinal mucosa and prevents villous atrophy, villous edema, and lacteal canal dilation. These results were obtained using rats with biliary obstruction.43 The lack of enterohepatic circulation during biliary obstruction also affects hepatic regeneration. The excretion rates of biliary lipids are disturbed with external biliary drainage compared to rates with internal biliary drainage, and liver regeneration rate is significantly correlated with bile flow and the excretion rates of biliary lipids.44 These results clearly indicate that the lack of enterohepatic circulation in the cholestatic liver is one of the major contributors to impaired hepatic regeneration.4
One of the most interesting targets of experimental investigation that has been undertaken so far has been the mechanism underlying cholestatic liver damage. Liver fibrosis is a complex phenomenon, in which nonparenchymal cells, such as Kupffer cells and HSCs, are reported to play a major role.45 Kupffer cells stimulate the progression of the fibrogenic process and HSCs synthesize most of the matrix proteins. Hepatic fibrosis develops 2–3 weeks after bile duct ligation in rats; this was the length of our experimental period. Mitochondrial oxidative stress and lipid peroxidation are considered to be the significant molecular players in cholestatic liver injury.46 A increase in oxidative over antioxidative biochemical activities, (i.e., during oxidative stress), has been reported to occur in liver fibrosis.1 Since lipids are a major target of oxidative stress, products of lipid peroxidation seem to play a role in cholestatic liver injury. The extent of oxidative stress is thought to be a crucial factor determining various pathophysiological conditions, with the spectrum of conditions ranging from cell modulation to fibrosis, to inflammation, irreversible cell damage, and apoptosis.45 It has been observed that in chronic cholestasis, an increased concentration of biliary acids in hepatocytes induces mitochondrial toxicity and results in the overproduction of ROS.47 Furthermore, increased lipid peroxidation has been reported to occur in the model of bile duct ligation.48 A statistically significant correlation between total bilirubin levels and lipid peroxidation has also been described in hepatic toxicity and diseases of the biliary tract.49 The observation of increased lipid peroxide levels in the blood of children with chronic cholestasis supports the oxidative hypothesis and explains, at least in part, the cholestatic-induced hepatocellular injury.50 Sokol et al. has also shown that exposure to hydrophobic bile acids causes a significant peroxidation of membrane lipids in isolated hepatocytes and liver mitochondria.51 In obstructive jaundice, increased hepatic lipid peroxidation, hepatocellular mitochondrial dysfunction, and decreased glutathione levels occur.52
Previous reports have shown that bile duct ligation in the rat for four weeks produces cirrhosis and a sixfold increase in liver collagen content.53 If the large biliary duct is obstructed for several weeks to months, as in our experiment, cholestasis extends throughout the lobule and hepatocyte necrosis leads to lobular bile infarcts. The ductular reaction develops at the periphery of the portal tracts and extends toward the neighboring portal tracts and deep into the parenchyma. This ductular reaction is accompanied by the proliferation of periductular (myo)fibroblasts that are derived from the portal fibroblasts around the interlobular bile ducts. Extensive fibrosis surrounding the ductular reaction enlarges the portal tracts and leads to the formation of porto-portal septa.2 In the septa development process of portal fibrogenesis, researchers maintain that viral hepatitis leads to the formation of porto-central septa, featuring activation of HSC, and that cholestatic liver diseases lead to portal–portal septa laid down by portal-tract-derived mesenchymal cells.54
During cirrhosis, the normal architecture of the liver is completely modified by the development of fibrotic septa. The cell populations involved differ according to their pattern of fibrosis. In portal diseases, such as chronic viral hepatitis and chronic bile duct obstruction, portal fibroblasts are found in the septa and HSCs are found at the interface between the septa and parenchyma.2 In the model of cholestatic fibrosis induced by common bile duct ligation, the withdrawal of intraductal pressure is sufficient to induce apoptosis in bile duct epithelial and portal fibroblastic cells, and to elicit extracellular matrix remodeling.55 These data provide insight into the mechanisms involved in the regression of cholestatic diseases and highlight the role of mechanical stimuli in cell growth and death.
The experimental bile duct ligation in rats, as in our experiment, is still one of the main models that support a link between oxidative damage and fibrogenesis during chronic cholestasis, which mimics clinical and morphological aspects of fibrosis and cirrhosis secondary to extrahepatic biliary obstruction in humans. We used 21 days of obstructive jaundice, as the collagen deposition was reported to become significant only after 15 days of bile duct ligation.23 The histopathological evaluation of liver tissue from the jaundiced group exhibited marked features of hepatocellular injury and fibrosis. In bile duct-ligated rats, the presence of marked oxidative stress and oxidative damage to lipids is traditionally demonstrated with thiobarbituric acid (TBA) reactive substance assays, which detect MDA, a peroxidation product of polyunsaturated fatty acids.56 MDA is considered to be the major product of lipid peroxidation.57 An MDA assay is generally used to detect the existence of lipid peroxidation as a result of oxidative stress in any tissue. In our study, we found that 21 days of obstructive jaundice resulted in a significant elevation of the concentration of liver MDA, compared with the sham-operated group (35.07 ± 3.73 vs. 20.86 ± 4.69 nmol/gr liver, p < 0.01; Table 2). This result is also in agreement with other studies on obstructive jaundice conducted in animals and humans.20 Moreover, the level of the antioxidant enzyme SOD, which is the scavenger of ROS in oxidative stress, was found to be significantly reduced in the jaundiced group (1.62 ± 1.36 vs. 7.86 ± 1.42 nmol/gr liver, p < 0.01; Table 2).
One of the major and early lipid peroxidation products is oxLDL. As the unsaturated fatty acids oxidize, they undergo molecular rearrangements and generate peroxy-fatty acids. These readily decompose, particularly in the presence of redox metals, such as iron and copper, to generate aldehydes, such as MDA, 4-hydroxynonenal, and others.58 During oxidative stress, plasma LDL is oxidatively modified mainly in the subendothelial space, attracting blood monocytes to the subendothelial spaces. These monocytes differentiate into macrophages, which further contribute to LDL oxidation. OxLDL is taken up by macrophages via scavenger receptors.59 These scavenger receptors have been shown to exist in many cells, including Kupffer cells, HSCs, macrophages, and liver endothelial cells.1,25,60 These phagocytic cells are reportedly responsible for a marked increase in the steady-state concentration of fibrogenic cytokines that intensify the cross-talk with extracellular matrix-producing cells.1 The liver is the primary organ for the clearance of the oxidatively modified cytotoxic and atherogenic LDL molecules. The effective filtration of oxLDL has been confirmed by the complete removal of these molecules from circulation within a few minutes when administered exogenously.60 Fluorescently-labeled oxLDL was essentially found to concentrate in Kupffer cells and, to a lesser extent, in endothelial cells in humans and in the rat liver. Following the rapid hydrolysis of cholesteryl esters from oxLDL by Kupffer cells, the molecule is transported to liver parenchymal cells and bile.61 Complete bile duct ligation may preclude the residual oxLDL transfer to the bile. Therefore, it is expected that oxLDL would accumulate in the liver after bile duct ligation. Previously, we have shown, using fluorescent immunostaining, that oxLDL accumulates in the liver of BALB/c mice after 10 days of experimental cholestasis.26 In this study, we used a prolonged period of obstructive jaundice, 21 days, as it has been demonstrated that, at high degrees of oxidation of LDL, the affinity for the oxLDL specific binding site is strongly enhanced and the phagocytic capacity of Kupffer cells increases within 2–3 weeks of biliary obstruction.62 Ongoing oxidative stress in prolonged cholestasis induces more ROS production than usual, which may enhance the oxidative modification of LDL within the liver. Free radicals not only induce oxidative stress in the liver but also initiate and maintain the oxidation of LDL.63 It has been demonstrated that there is a close relationship between the severity of fibrosis and the amount of lipid peroxidation products, which reflects the level of oxidative stress.64 Moreover, there is a close link between lipid peroxidation and the activation of inflammatory cells, and a positive linear correlation exists between collagen deposition and the production of MDA.23 Our results related to the MDA concentration are in agreement with the literature.23,64
Independently from the nature of the injury, the fibrogenic pathway is similar in various tissues. Kupffer cells and HSCs are the key players in liver fibrosis. The phenotypic transformation of HSCs into MFB-like cells is considered to be the central event of hepatic fibrogenesis. The activation of HSCs results in the production of a large quantity of extracellular matrix components. In an in vitro study, it was shown that there were specific receptors for oxLDL in HSCs that stimulate the synthesis of the extracellular matrix, a result that is consistent with the hypothesis that oxLDL might at least participate in the events leading to liver inflammation and fibrosis in cholestasis.25
In conclusion, we have demonstrated that oxLDL accumulates in liver tissue sections of prolonged obstructive jaundiced rats. Furthermore, we show that there is significant hepatic fibrosis evident upon histopathological examination. The accumulation of oxLDL may be the consequence of either a failure to eliminate it via bile flow or a cholestasis-induced enhancement of ROS production and lipid peroxidation, which may predict ongoing oxidative stress. Oxidative damage, possibly in association with more than one mechanism, appears to be crucial for the progression of fibrosis through cirrhosis in cholestatic liver injury. We believe that oxLDL might at least play a contributory role in the various pathophysiological mechanisms underlying the process of liver fibrosis and that the association of oxLDL accumulation and hepatic fibrosis deserves to be considered as something more than coincidence in cholestasis. Further studies are needed to evaluate the potential effects of oxLDL in the progression to secondary biliary cirrhosis.