




This study aimed to identify novel PITX2c mutations responsible for idiopathic atrial fibrillation.
METHODS:A cohort of 210 unrelated patients with idiopathic atrial fibrillation and 200 unrelated, ethnically matched healthy individuals used as controls were recruited. The whole coding exons and splice junctions of the PITX2c gene, which encodes a paired-like homeobox transcription factor required for normal cardiovascular morphogenesis, were sequenced in 210 patients and 200 control subjects. The causative potentials of the identified mutations were automatically predicted by MutationTaster and PolyPhen-2. The functional characteristics of the PITX2c mutations were explored using a dual-luciferase reporter assay system.
RESULTS:Two novel heterozygous PITX2c mutations (p.Q105L and p.R122C) were identified in 2 of the 210 unrelated patients with idiopathic atrial fibrillation. These missense mutations were absent in the 400 control chromosomes and were both predicted to be pathogenic. Multiple alignments of PITX2c protein sequences across various species showed that the altered amino acids were highly evolutionarily conserved. A functional analysis demonstrated that the mutant PITX2c proteins were both associated with significantly reduced transcriptional activity compared with their wild-type counterparts.
CONCLUSION:The findings of this study associate PITX2c loss-of-function mutations with atrial fibrillation, supporting the hypothesis that dysfunctional PITX2c confers enhanced susceptibility to atrial fibrillation and suggesting potential implications for early prophylaxis and allele-specific therapy for this common arrhythmia.
Atrial fibrillation (AF), a supraventricular tachycardia characterized by rapid and chaotic atrial electrical activity with subsequent deterioration of atrial mechanical function, is the most prevalent form of sustained cardiac arrhythmia in humans worldwide and is responsible for approximately one-third of all hospital admissions with miscellaneous heart rhythm disturbances (1). The prevalence of AF is approximately 1% in the general population and increases strikingly with advanced age, increasing from <1% in individuals younger than 60 years to approximately 10% in subjects older than 80 years (1). According to the Framingham Heart Study, the lifetime risk for developing AF is approximately 25% for people older than 40 years (2). The prevalence of AF in the United States is currently estimated at 2.3 million and is projected to exceed 10 million by 2050 (3). This common arrhythmia significantly contributes to a degraded quality of life, reduced exercise capacity, cognitive impairment or dementia, tachycardiomyopathy, thromboembolism, congestive heart failure, and even death (1,4,5). The mortality of patients with AF is approximately twice that of age- and gender-matched patients with normal sinus rhythms, independent of pre-existing cardiovascular conditions (6). AF patients have a 2- to 7-fold increased risk for ischemic stroke compared with individuals without AF, and 15%–20% of all strokes are ascribed to AF (7). Note that the risk of thromboembolism attributable to AF also increases markedly with increased age, ranging from 1.5% among patients aged 50–59 years to 23.5% among patients aged 80–89 years (7). Consequently, AF imposes a substantial economic burden on health care systems because of its increased morbidity- and mortality-associated therapeutic interventions. This socioeconomic burden is anticipated to continually increase in the future as life expectancies increase (8). Despite its high prevalence and important clinical significance, the molecular basis of AF remains poorly understood.
Frequently, AF is associated with diverse structural heart diseases or systemic disorders, such as ischemic heart disease, valvular heart disease, congenital heart disease, pulmonary heart disease, cardiomyopathy, cardiothoracic surgery, congestive heart failure, essential hypertension, and hyperthyroidism (1,9). Other risk factors for AF include aging, obesity, sleep apnea, cigarette smoking, excessive alcohol consumption, and exposure to drugs or toxicants (1,10). However, in 12%–30% of all AF patients and 20%–45% of younger AF patients, AF occurs alone in the absence of the above-mentioned underlying diseases or precipitating factors; this condition is defined as idiopathic or lone AF and termed familial AF, as at least 15% of patients have positive family histories of AF (1,11). A growing number of epidemiological studies have demonstrated the familial aggregation of AF and the enhanced susceptibility to AF in the close relatives of AF patients, indicating that genetic risk factors play key roles in the pathogenesis of AF in a subset of patients (12). A whole-genome scan with polymorphic microsatellite markers and linkage analysis mapped the AF susceptibility loci on human chromosomes 10q22, 6q14-16, 11p15.5, 5p15, 10p11-q21, and 5p13, for which the AF-causing mutations in 2 genes, KCNQ1 on chromosome 11p15.5 and NUP155 on chromosome 5p13, were identified and functionally characterized (13). Analyses of the candidate genes revealed a long list of AF-associated genes, including KCNE2, KCNH2, KCNJ2, KCNA5, SCN5A, ANP, GJA5, GATA4, GATA5, GATA6, and NKX2-5 (13-21). Nevertheless, AF is genetically heterogeneous, and the genetic defects underlying AF in an overwhelming majority of patients remain to be identified.
Recently, a genome-wide association study identified 2 sequence variants (rs2200733 and rs10033464) on chromosome 4q25 that were strongly associated with an enhanced vulnerability for AF (22). This association was subsequently replicated in a study of 4 large populations with ambulatory AF (23) and was also reported for post-cardiac surgery AF in a setting thought to be related to inflammation (24). Moreover, the 2 sequence variants were observed to increase the risk for both the early and late recurrences of AF after catheter ablation (25) and acted as genetic modifiers of rare ion channel mutations associated with familial AF (26). The PITX2 gene is closest to these sequence variants; it a member of the pituitary homeobox (PITX) family of transcription factors, which play a pivotal role in embryonic morphogenesis. PITX2c is the predominant isoform that is expressed in embryonic and adult hearts (27). Emerging evidence underlines the essential role of PITX2c in the embryonic development of the left atrium, cardiac conduction system, and pulmonary venous myocardium, a major source of ectopic activity that is implicated in initiating and maintaining AF (28). The abnormal expression of PITX2c has been associated with an increased predisposition to AF (29-31). These findings justify screening for PITX2c as a prime candidate gene for idiopathic AF.
MATERIALS AND METHODSEthics statementThis study was performed in compliance with the ethical principles of the revised Declaration of Helsinki (Somerset West, Republic of South Africa, 1996). The research protocol was reviewed and approved by the local institutional ethics committee, and written informed consent was obtained from all participants prior to the study.
Study subjectsA cohort of 210 unrelated patients with idiopathic AF was recruited from the Han Chinese population. Patients with enlarged left atriums (≥40 mm in left atrial diameter) were excluded. A total of 200 ethnically matched, unrelated healthy individuals were enrolled as the controls. Peripheral venous blood samples were prepared, and clinical data, including medical records, electrocardiogram, and echocardiography reports, were collected. The study subjects were clinically classified using a consistently applied set of definitions (11,18). Briefly, the AF diagnosis was made using a standard 12-lead electrocardiogram demonstrating no P waves and irregular R-R intervals, regardless of the clinical symptoms. Idiopathic AF was defined as AF occurring in individuals without other cardiac or systemic diseases using a physical examination, electrocardiogram, transthoracic echocardiogram, or extensive laboratory tests. Subjects were classified as ‘healthy’ if they were asymptomatic and had normal electrocardiograms. In addition, paroxysmal AF was defined as AF lasting more than 30 seconds that terminated spontaneously. Persistent AF was defined as AF lasting more than 7 days and requiring either pharmacologic therapy or electrical cardioversion for termination. AF that was refractory to cardioversion or that was allowed to continue was classified as long-lasting persistent AF.
Genetic scanGenomic DNA was extracted from the peripheral venous blood lymphocytes of all participants with a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). The whole coding region and splice junction sites of PITX2c was sequenced in 210 unrelated patients with idiopathic AF, and the genotyping PITX2c in the 200 control individuals was performed subsequently to identify the presence of mutations in the patients. The referential genomic DNA sequence of PITX2c was derived from the GenBank (accession No. NC_000004), which is at the National Center for Biotechnical Information (NCBI, http://www.ncbi.nlm.nih.gov/). The primer pairs used to amplify the coding exons and exon-intron boundaries of PITX2c by polymerase chain reaction (PCR) were designed with the help of the online Primer 3 program (http://frodo.wi.mit.edu), as shown in Table1. PCR was performed using HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster City, CA, USA) under standard conditions and concentrations of reagents. Amplified products were analyzed on 1% agarose gels stained with ethidium bromide and purified with QIAquick Gel Extraction Kit (Qiagen). Both strands of each amplicon were sequenced with a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems). The sequencing primers were the same as those previously designed for specific region amplification. The DNA sequences were analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems). The variant was validated by re-sequencing an independent PCR-generated amplicon from the subject and meeting the quality control thresholds with a call rate >99%. In addition, for an identified sequence variant, the Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS) and NCBI's single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) online databases were searched to confirm its novelty.
Primers to amplify the coding exons and exon-intron boundaries of PITX2c.
| Exon | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) | Size (bp) |
|---|---|---|---|
| 1 | CAG,CTT,GGC,TTG,AGA,ACT,CG | TGA,CTT,CCT,TGG,GGC,GAG,AG | 442 |
| 2 | CAG,CTC,TTC,CAC,GGC,TTC,TG | GCT,GCC,TTC,CAC,ATT,CTC,TC | 387 |
| 3 | AAT,CTG,CAC,TGT,GGC,ATC,TG | AGT,CTT,TCA,AGG,GCG,GAG,TT | 677 |
Multiple PITX2c protein sequences across various species were aligned using the online program MUSCLE, version 3.6 (http://www.ncbi.nlm.nih.gov/).
Prediction of the disease-causing potential of a PITX2c sequence variationThe disease-causing potential of a PITX2c sequence variation was predicted using MutationTaster (an online program at http://www.mutationtaster.org), which automatically provides a probability for the variation to be either a pathogenic mutation or a benign polymorphism. Note that the p-value used here is the probability of the correct prediction rather than the probability of error, as used in t-test statistics (i.e., a value close to 1 indicates a high accuracy of the prediction). Another online program, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), was also used to evaluate the mutational pathogenicity.
Expression plasmids and site-directed mutagenesisThe recombinant expression plasmid PITX2c-pcDNA4, which was constructed by M. Hermina Strungaru et al. (32), was a gift from Prof. Georges Christé, Physiopathologie des Troubles du Rythme Cardiaque, Faculté de Pharmacie de Lyon, Université Lyon 1, Lyon, France. The atrial natriuretic factor (ANF)-luciferase reporter plasmid, which contains the 2600-bp 5'-flanking region of the ANF gene, namely ANF (-2600)-Luc, was kindly provided by Dr. Ichiro Shiojima, from the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chuo-ku, Chiba, Japan. Each of the identified mutations was introduced into the wild-type PITX2c using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) with a complementary pair of primers. The mutants were sequenced to confirm the desired mutations and to exclude any other sequence variations.
Luciferase reporter gene assayChinese hamster ovary (CHO) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. CHO cells were grown 24 h prior to the transfection. The ANF(-2600)-Luc reporter construct and an internal control reporter plasmid pGL4.75 (hRluc/CMV, Promega) were used in transient transfection assays to explore the transactivational activity of the PITX2c mutants. The CHO cells were transfected with 2 μg of wild-type PITX2c–pcDNA4, mutant PITX2c–pcDNA4 (Q105L or R122C), or empty vector pcDNA4, 2.0 μg of ANF(-2600)-Luc reporter construct, and 0.04 μg of pGL4.75 control reporter vector using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA). For the co-transfection experiments, 1 μg of wild-type PITX2c–pcDNA4, 1 μg of mutant PITX2c–pcDNA4 (Q105L or R122C), 2.0 μg of ANF(-2600)-Luc, and 0.04 μg of pGL4.75 were used. The transfected cells were incubated for 24 h, and then they were lysed and assayed for the reporter activities. Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega). The activity of the ANF promoter was presented as the fold activation of Firefly luciferase relative to the Renilla luciferase. A minimum of three independent experiments were performed for wild-type or mutant PITX2c.
Statistical analysisThe data are expressed as the means±SD. Continuous variables were tested for normality of distribution, and Student′s unpaired t-test was used to compare the numeric variables between 2 groups. The categorical variables were compared between 2 groups using Pearson's chi-squared test or Fisher′s exact test, as appropriate. A 2-sided p-value <0.05 was considered statistically significant.
RESULTSClinical characteristics of the study populationA cohort of 210 unrelated patients with idiopathic AF were enlisted, clinically evaluated, and compared with 200 ethnically matched, unrelated healthy individuals. None of the participants had the traditional risk factors for AF. There were no significant differences between the patient and control groups in the baseline characteristics, including age, gender, body mass index, blood pressure, fasting blood glucose, serum lipid, left atrial dimension, left ventricular ejection fraction, heart rate at rest, and lifestyle (data not shown). The baseline clinical characteristics of the 210 patients with idiopathic AF are summarized in Table2.
Baseline clinical characteristics of the study subjects.*)
| Clinical characteristics | Patient group (n = 210) | Control group (n = 200) |
|---|---|---|
| Male (%) | 96 (46) | 92 (46) |
| Age at the initial AF diagnosis (years) | 53.2±8.7 | NA |
| Age at the time of the study (years) | 56.7±10.1 | 58.3±9.5 |
| Type of AF at presentation | ||
| Paroxysmal AF (%) | 147 (70) | 0 (0) |
| Persistent AF (%) | 38 (18) | 0 (0) |
| Long-lasting persistent AF (%) | 25 (12) | 0 (0) |
| Positive family history of AF (%) | 32 (15) | 0 (0) |
| History of cardioversion (%) | 178 (85) | 0 (0) |
| Implanted cardiac pacemaker (%) | 6 (3) | 0 (0) |
| Resting heart rate (beats per minute) | 76.5±11.8 | 77.2±10.5 |
| Systolic blood pressure (mmHg) | 130.4±12.6 | 131.0±13.3 |
| Diastolic blood pressure (mmHg) | 85.8±7.3 | 86.2±8.1 |
| Body mass index (kg/m2) | 22.7±2.0 | 23.0±2.4 |
| Left atrial dimension (mm) | 38.2±3.6 | 37.5±3.9 |
| Left ventricular ejection fraction (%) | 62.8±7.2 | 63.4±6.7 |
| Fasting blood glucose (mmol/L) | 4.5±0.4 | 4.6±0.5 |
| Total cholesterol (mmol/L) | 4.2±0.3 | 4.1±0.4 |
| Triglycerides (mmol/L) | 1.6±0.2 | 1.5±0.3 |
| Medications | ||
| Amiodarone (%) | 160 (76) | 0 (0) |
| Warfarin (%) | 151 (72) | 0 (0) |
| Digoxin (%) | 44 (21) | 0 (0) |
| Beta-blocker (%) | 17 (8) | 0 (0) |
| Calcium channel blocker (%) | 10 (5) | 0 (0) |
* There were no significant differences in the baseline characteristics between the patient and control groups.
NA indicates not applicable or not available.
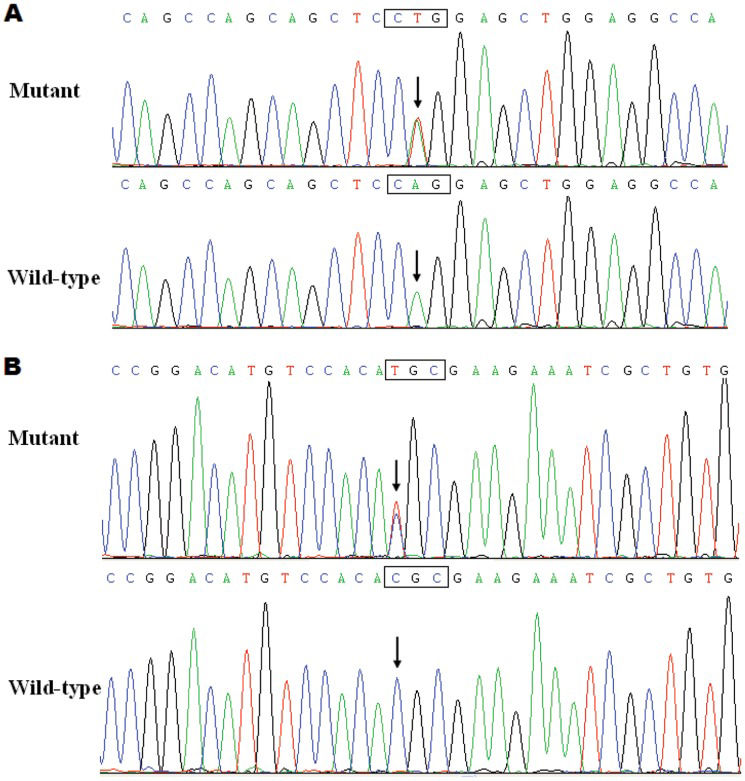
Two heterozygous missense mutations in PITX2c were identified in 2 of the 210 unrelated patients with idiopathic AF. The total population prevalence of PITX2c mutations based on the patient cohort was approximately 0.95%. Specifically, a substitution of thymine (T) for adenine (A) in the second nucleotide of codon 105 (c.314A>T), which predicted the transition of glutamine (Q) into leucine (L) at amino acid 105 (p.Q105L), was identified in a 52-year-old female patient who was initially diagnosed with AF at the age of 35 years. A change of cytosine (C) into thymine (T) in the first nucleotide of codon 122 (c.364C>T), corresponding to the transversion of arginine (R) into cysteine (C) at amino acid 122 (p.R122C), was found in a 48-year-old male patient who was first diagnosed with AF at the age of 21 years. The 2 mutation carriers had no apparent malformations in the eyes, teeth, umbilicus, or heart and had no positive family history. No relatives from these 2 mutation carriers were available for PITX2c genotyping. The sequence chromatograms showing the detected heterozygous PITX2c mutations of c.314A>T and c.364C>T in contrast to their control sequences are shown in Figures1A and 1B), respectively. A schematic diagram of PITX2c showing the structural domains and the locations of the identified mutations is presented in Figure2 (33,34). These missense mutations were neither observed in the control population nor reported in the EVS and NCBI SNP databases.
, mutant) or C/T (Figure1B), mutant) or the homozygous nucleotides of A/A (Figure1A), wild-type) or C/C (Figure1B), wild-type). The rectangle designates the nucleotides comprising a codon of PITX2c.")
Sequence electropherograms showing the PITX2c mutations in contrast with their corresponding controls. The arrows indicate the heterozygous nucleotides of A/T (Figure1A), mutant) or C/T (Figure1B), mutant) or the homozygous nucleotides of A/A (Figure1A), wild-type) or C/C (Figure1B), wild-type). The rectangle designates the nucleotides comprising a codon of PITX2c.
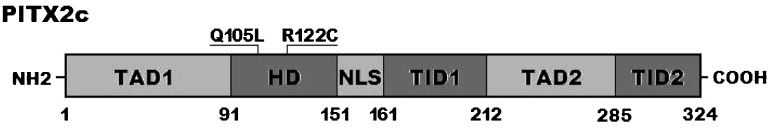
; HD, homeodomain (amino acids 92–151); NLS, nuclear localization signal (amino acids 145–161); TID1, transcriptional inhibitory domain 1 (amino acids 162–212); TAD2, transcriptional activation domain 2 (amino acids 213–285); TID2, transcriptional inhibitory domain 2 (amino acids 286–324); and COOH, carboxyl-terminus.")
A schematic representation of the PITX2c protein structure with the atrial fibrillation-related mutations indicated. The mutations identified in patients with atrial fibrillation are shown above the structural domains. NH2 means amino-terminus; TAD1, transcriptional activation domain 1 (amino acids 1–91); HD, homeodomain (amino acids 92–151); NLS, nuclear localization signal (amino acids 145–161); TID1, transcriptional inhibitory domain 1 (amino acids 162–212); TAD2, transcriptional activation domain 2 (amino acids 213–285); TID2, transcriptional inhibitory domain 2 (amino acids 286–324); and COOH, carboxyl-terminus.
The alignment of PITX2c protein sequences across 11 species ranging from humans to mammals and insects showed that the altered amino acid residues were completely (for p.R122) or highly (for p.Q105) evolutionarily conserved among PITX2c orthologs, which suggested that these particular arginine and glutamine residues in the homeodomain of PITX2c are functionally important (Figure3).
Disease-causing potential of the PITX2c variations
The PITX2c sequence variations of c.314A>T and c.364C>T were both automatically predicted by MutationTaster to be disease-causing mutations with the same p-value of 1.0. No SNP in the altered regions was found in the MutationTaster database. The 2 PITX2c sequence variations were also predicted by PolyPhen-2 to most likely be damaging, with scores of 0.999 for c.314A>T (sensitivity 0.14; specificity 0.99) and 1.000 for c.364C>T (sensitivity 0.00, specificity 1.00).
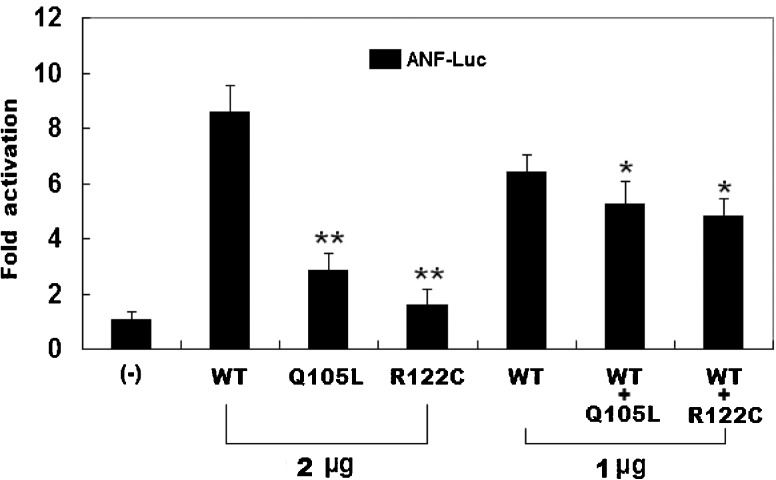
Transcriptional activity of the PITX2c mutantsThe same amounts of wild-type (2 μg), Q105L-mutant (2 μg), and R122C-mutant PITX2c (2 μg) activated the ANF promoter by ∼9-fold, ∼3-fold, and ∼2-fold increases, respectively, compared with the empty plasmid. When the same amount of wild-type PITX2c (1 μg) was cotransfected with Q105L-mutant PITX2c (1 μg) or R122C-mutant PITX2c (1 μg), the induced activation of the ANF promoter was a ∼5-fold increase, compared with the empty plasmid (Figure4).
 promoter-driven luciferase reporter in the CHO cells by PITX2c wild-type (WT), Q105L-mutant, or R122C-mutant, alone or in combination, demonstrated a significantly decreased transactivational activity by the mutant proteins. The experiments were performed in triplicate, and the means and standard deviations are shown. ** indicates p<0.001 and * denotes p<0.01, when compared with the same amount (2 μg) of wild-type PITX2c.")
The functional defects associated with the PITX2c mutations. The activation of an atrial natriuretic factor (ANF) promoter-driven luciferase reporter in the CHO cells by PITX2c wild-type (WT), Q105L-mutant, or R122C-mutant, alone or in combination, demonstrated a significantly decreased transactivational activity by the mutant proteins. The experiments were performed in triplicate, and the means and standard deviations are shown. ** indicates p<0.001 and * denotes p<0.01, when compared with the same amount (2 μg) of wild-type PITX2c.
In the present study, 2 novel heterozygous PITX2c mutations, p.Q105L and p.R122C, were identified in 2 unrelated patients with idiopathic AF; these mutations were absent in the 400 reference chromosomes and were both predicted to be pathogenic by MutationTaster and PolyPhen-2. A cross-species alignment of PITX2c protein sequences showed that the altered amino acids are highly evolutionarily conserved. The functional analysis demonstrated that the mutant PITX2c proteins are associated with significantly decreased transcriptional activity. Therefore, it is likely that having functionally impaired PITX2c predisposes a patient to AF.
PITX2 is expressed as several protein isoforms, which are generated by differential mRNA splicing and alternative translation initiation sites. In the human and the mouse, the transcriptionally functional isoforms are PITX2-a, -b, and -c, which share an identical homeodomain and carboxyl terminus but differ in their amino termini. Humans possess an additional isoform, the D isoform, which lacks the amino-terminal domain and most of the homeodomain (31). PITX2c is the major isoform that is expressed asymmetrically in the developing and adult heart and plays a crucial role in normal cardiovascular genesis and maturation (30,31). The human PITX2c gene maps to chromosome 4q25, which consists of 3 exons coding for a protein of 324 amino acids (35). One of the most important functional domains of PITX2c is the homeodomain that recognizes and binds to the specific consensus DNA sequence 5′-TAATCC-3′. This homeodomain is essential for DNA binding, nuclear translocation, and interaction with other transcription factors (35). The PITX2c mutations of p.Q105L and p.R122C, which are identified in the present study, are both located in this homeodomain; thus, they may be expected to influence the transcriptional activity of PITX2c by interfering with its DNA-binding ability.
Previous studies have corroborated that PITX2c is an upstream regulator of multiple target genes that are expressed in the heart during embryogenesis, including the gene that encodes ANF (36). Therefore, the functional role of a PITX2c mutation can be characterized by an assay of the transcriptional activity of the ANF promoter in the cells expressing PITX2c mutants, in contrast to the wild-type counterpart. In this study, the functional characteristics of the 2 novel PITX2c mutations identified in the AF patients were delineated by a transcriptional activity analysis, the results of which showed that both mutations were associated with a significantly reduced transcriptional activity on a downstream gene. This result suggests that the dysfunctional PITX2c resulting from mutations is potentially an alternative pathological mechanism in AF.
The finding that functionally impaired PITX2c contributes to AF may be partially attributed to the abnormal development of cardiovascular system, especially pulmonary venous myocardium (37,38). PITX2c is abundantly expressed in the atria and pulmonary myocardium, downregulating the sinoatrial nodal gene program, for example, Shox2, HCN4 and Cav3.1, and upregulating a gene program characteristic of a working myocardium phenotype, for example, Nkx2.5, Cx40, Cx43, ANP, and Kir2.1 (27,29,31,37). Therefore, PITX2c loss-of-function mutations presumably predispose a patient to AF by inducing the identity switch of the atrial and pulmonary myocardium to a sinoatrial node–like phenotype, thereby forming an electrophysiological substrate that favors AF. Another equally compelling explanation is that dysfunctional PITX2c may change the expression profile of ion channels in the atrium, including an increased expression of the potassium-channel gene KCNQ1, which alters atrial repolarization, as suggested by the gene expression analysis and functional studies in PITX2-deficient mice (29,31,39).
It has been shown that some downstream genes are transactivated by PITX2c (27), and mutations in multiple target genes, including Nkx2.5, Cx40, Cx43, and ANP, have been causally implicated in AF (14,21,40-42). This result implies that mutated PITX2c may confer vulnerability to AF by downregulating the expressions of these target genes. Therefore, additional experiments investigating whether PITX2c mutations can modify the expression levels of some of the genes involved in the AF pathogenesis are still required.
Notably, PITX2 mutations were previously implicated in type 1 Axenfeld-Rieger syndrome, type 2 iridogoniodysgenesis, Peters' anomaly, ring dermoid of cornea, and congenital cardiac malformation (33,43-45). In this study, the mutations identified in the AF patients were in the homeodomain shared by PITX2a, PITX2b, and PITX2c, which implies that mutated PITX2 may also be responsible for AF.
In addition, Yang et al. (46) previously screened 152 index patients with familial AF who were enlisted from the Han Chinese population. They identified 2 novel heterozygous PITX2c mutations, including c.110C>G, corresponding to p.S37W, and c.840 T>A, which resulted in p.Y280X in 2 of the 152 AF probands, with a mutational prevalence of approximately 1.32%. The authors' analysis of the pedigrees showed that each mutation that co-segregated with AF was transmitted in an autosomal dominant manner in the family, with complete penetrance. However, the functional characteristics of the PITX2 mutations associated with familial AF remain to be addressed.
In conclusion, this study links PITX2c loss-of-function mutations to AF for the first time, provides evidence that functionally impaired PITX2c is associated with an increased vulnerability to AF, and identifies the potential implications for early prophylaxis and allele-specific therapies for this common arrhythmia.
ACKNOWLEDGMENTSWe thank the study participants for their devotion to the study. This work was partially supported by grants from the National Natural Science Fund of China (81070153, 81270161 and 30570768), the National Basic Research Program of China (2010CB912604), and the Personnel Development Foundation of Shanghai, China (2010019).
AUTHOR CONTRIBUTIONSQiu XB contributed to the experimental design, the clinical and experimental research, the data analysis and interpretation, and the manuscript writing. Xu YJ, Li RG, Xu L, Liu X, and Fang WY contributed to the clinical research and the data analysis and interpretation. Yang YQ and Qu XK contributed to the study design, the experimental research, the data analysis and interpretation, and the initial drafting and revision of the manuscript.
No potential conflict of interest was reported.