



Analizar la relación entre las cardiopatías congénitas (CC) y las cromosomopatías en vida fetal.
MétodoEstudio retrospectivo realizado en un centro terciario de referencia. Seleccionamos las CC diagnosticadas prenatalmente entre 1990 y 2011, con verificación posnatal del diagnóstico y con información disponible del cariotipo. La recomendación de realizar técnica invasiva prenatal para estudio del cariotipo dependió del tipo de CC y de la existencia de otros factores de riesgo de cromosomopatía.
ResultadosSe analizaron 1.384 CC. El cariotipo se estudió prenatalmente en 848 (61,3%) y en el resto o se estudió posnatalmente (172; 12,4%) o se excluyó clínicamente la presencia de cromosomopatía por la ausencia de marcadores clínicos indicativos de aquella (364; 26,3%). Existía una cromosomopatía en 363 CC (26,2%). El diagnóstico fue prenatal en 324 (89,3%) y posnatal en 39 (10,7%). En estos casos no se realizó el estudio prenatal del cariotipo principalmente por la negativa de los padres (n=28). La CC que mostró mayor asociación con cromosomopatías fue el canal aurículo-ventricular (66,7%). Esta asociación fue nula en algunas CC como la transposición de grandes arterias o el ventrículo único. Lo mismo sucedió en la atresia tricúspide aislada y en los síndromes de heterotaxia sin anomalías ajenas a las que forman parte del síndrome.
ConclusionesAun siendo enormemente relevante la información del cariotipo en los fetos con CC para la toma de decisiones de los padres y el pronóstico del paciente, la recomendación de dicho estudio ha de individualizarse según las características de cada caso, pudiendo evitarse los riesgos de la técnica invasiva diagnóstica en muchos casos.
To assess the relationship between congenital heart defects (CHD) and chromosomal abnormalities in fetal life.
MethodsThis is a retrospective study undertaken at a tertiary care referral center. Our database was queried for cases of CHD prenatally diagnosed between 1990 and 2011, with postnatal diagnostic verification, as well as information available as regards the karyotype. The recommendation for performing fetal invasive procedures relied upon the type of CHD and the presence of associated high-risk factors of chromosomal disease.
ResultsA total of 1,384 CHD were retrieved and analyzed. The karyotype was studied prenatally in 848 (61.3%) and in the rest was either studied postnatally (172, 12.4%) or the presence of chromosomal disease was clinically ruled out given the absence of suggestive clinical markers (364, 26.3%). Chromosomal defects were diagnosed in 363 CHD (26.2%). The diagnosis was made prenatally in 324 (89.3%), and after birth in 39 (10.7%). In most of these cases (n=28) the parents refused fetal invasive testing. We found that atrioventricular septal defect was the CHD most associated with chromosomal abnormalities (66.7%). On the contrary, we did not observe any chromosomal defect in CHD, such as transposition of large arteries or single ventricle. Similarly, there was no abnormal karyotype in isolated tricuspid atresia or in heterotaxy syndromes presenting without anomalies other than those typically included in the disease.
ConclusionsKaryotype analysis is highly relevant in fetuses with CHD, given its impact in the parental decision-making process and patient outcome. Nevertheless, the recommendation of performing fetal invasive testing should be based on the individual characteristics of any given case, and in many cases the risks associated with the invasive procedure could be avoidable.
Las cardiopatías congénitas (CC) constituyen las malformaciones congénitas severas más frecuentes y su prevalencia estimada es de 8–12 casos/1.000 nacidos vivos1–3. Son las responsables del 20-30% de las muertes neonatales y del 50% de la mortalidad infantil debida a anomalías congénitas4–6.
La mayoría de las CC que son detectadas en vida fetal corresponden a las formas más severas. Además, la incidencia de anomalías cromosómicas asociadas y síndromes genéticos es alta, especialmente si junto a la CC coexisten otras malformaciones, lo cual contribuye a empeorar el pronóstico de la CC, aumentando el riesgo de aborto espontáneo, muerte fetal y neonatal7–11. En efecto, el riesgo de anomalía cromosómica acompañando a una CC diagnosticada prenatalmente se estima en un 15-25%9 mientras que en las series pediátricas esta asociación se presenta en el 5-10%7. Esta discrepancia puede ser explicada, al menos en parte, por la mayor tasa de pérdidas fetales cuando concurren CC y cromosomopatía. Por todo ello, clásicamente se ha ofrecido de forma sistemática el estudio del cariotipo en todo feto diagnosticado de CC. Sin embargo, la probabilidad de que en un determinado feto con CC se diagnostique una cromosomopatía es claramente dependiente del tipo de CC, siendo extremadamente improbable esta asociación en determinadas CC11. Si a esto le añadimos que para conocer el cariotipo fetal debemos someter el embarazo a una técnica invasiva no exenta de riesgos, cabe preguntarse si en la actualidad y a la luz de los conocimientos existentes se puede individualizar la recomendación de la realización del cariotipo del feto en función de las características de la CC.
El objetivo de este artículo es, a partir del estudio de nuestra propia casuística, analizar la relación existente entre las diferentes CC y las cromosomopatías.
MétodosEste es un estudio longitudinal retrospectivo realizado en un centro terciario de referencia para el diagnóstico prenatal y el tratamiento de patología fetal y neonatal. Se han analizado todas las CC estructurales y/o posicionales diagnosticadas prenatalmente entre 1990 y 2011 cuyo diagnóstico fue verificado posnatalmente, y con estudio del cariotipo disponible. Se excluyeron los casos en los que no se produjo confirmación diagnóstica posnatal por no contar con la necropsia después de la muerte fetal, neonatal o tras la interrupción voluntaria del embarazo (IVE) y aquellos casos en los que no se disponía de seguimiento. También fueron excluidas las miocardiopatías, los tumores cardíacos y los trastornos del ritmo salvo que coexistieran con un defecto congénito cardíaco estructural o posicional. Para su clasificación, los fetos con CC en que hubiera varios defectos anatómicos fueron incluidos en la categoría correspondiente a la lesión dominante.
En cada caso se procedió de forma sistemática a la realización de una exploración ecográfica detallada incluyendo ecocardiografía fetal ampliada en modo-2D, con estudio doppler color y pulsado, así como una ecografía morfológica para excluir otras anomalías extracardíacas asociadas. Todas las exploraciones ecográficas se realizaron por un experto en medicina fetal y con equipos ecográficos de alta calidad, preferentemente por vía transabdominal, aunque esta se complementó en algunos casos con ecografía transvaginal, especialmente en el primer trimestre de la gestación. Todos los casos fueron grabados en vídeo para su posterior análisis. En los casos diagnosticados ≥14 semanas se revisó la información relativa al valor de la translucencia nucal (TN). Valores de esta >p95 se consideraron anormales12.
Si al realizar el diagnóstico prenatal de la CC no se disponía de estudio previo del cariotipo, la recomendación de realizar técnica invasiva para proceder a dicho estudio dependió del tipo de CC y de la existencia o no de otros factores de riesgo de cromosomopatía tales como la presencia de anomalías extracardíacas. En ausencia de factores asociados de riesgo y en CC tales como la transposición de grandes arterias (TGA), transposición congénitamente corregida (TccGA), síndromes de heterotaxia, hipoplasia de ventrículo izquierdo en sus formas clásicas (atresia aórtica, atresia mitro-aórtica, estenosis aórtica crítica evolucionada a ventrículo izquierdo hipoplásico), estenosis pulmonar y ventrículo único (VU), no se ofreció sistemáticamente el estudio del cariotipo fetal. En caso de realizar técnica invasiva, el tipo de técnica dependió básicamente de la edad gestacional (EG) al diagnóstico, reservando la funiculocentesis habitualmente para aquellos casos diagnosticados ≥20 semanas. En aquellos casos en que al nacimiento no se disponía de estudio del cariotipo fetal, la decisión pediátrica de realizar dicho estudio dependió también del tipo de CC y de los hallazgos clínicos o de imagen existentes en cada caso que pudieran hacer sospechar la existencia de una anomalía cromosómica. El estudio de la microdeleción 22q11, tanto prenatal como posnatalmente, se incluyó en la sistemática de estudio del cariotipo fetal en la mayoría de las CC conotruncales a partir de 1999.
La mayoría de los casos, y principalmente los diagnosticados en los primeros años del estudio, fueron analizados conjuntamente con un cardiólogo infantil. Tras el diagnóstico prenatal los padres fueron informados de las implicaciones de la CC en cuanto al pronóstico y tratamiento, así como del riesgo de recurrencia. Tras el asesoramiento, en aquellos casos en que los padres optaron por la IVE, se les aconsejó sobre la necesidad de realizar la autopsia para confirmar los hallazgos ecográficos y excluir otras anomalías estructurales no detectadas prenatalmente. En aquellos casos en los que los padres decidieron continuar con la gestación se ofreció un seguimiento ecográfico cada 4-6 semanas hasta el parto con el fin de monitorizar el crecimiento y bienestar fetal, detectar posibles anomalías asociadas y evaluar la progresión de la CC. En la mayoría de las CC graves, entendidas por tales aquellas que requieren cirugía en el primer año de vida13 se recomendó que el parto tuviera lugar en centro terciario con disponibilidad de cuidados intensivos neonatales avanzados, cardiología infantil y cirugía cardíaca infantil. En los casos en que se produjo la muerte antenatal o neonatal también se ofreció estudio post-mortem y asesoramiento clínico a los padres tras el estudio.
La actitud obstétrica se rigió por criterios convencionales en la inmensa mayoría de los casos y solo si existía fracaso cardíaco severo se optó por una cesárea electiva por la CC fetal. Tras el nacimiento se procedió a la realización de una valoración clínica general y cardiológica por el neonatólogo y el cardiólogo pediátrico, y una ecocardiografía para confirmar o descartar la CC, adaptando posteriormente la conducta a las características de cada caso.
Los datos de seguimiento fueron recogidos a partir del análisis de las bases de datos informatizadas de la Unidad de Medicina Fetal, del Servicio de Obstetricia y Ginecología y del Servicio de Neonatología. Los resultados de los estudios citogenéticos fueron incluidos prospectivamente en la ficha de cada paciente tan pronto como fueron proporcionados por el servicio de Genética. En cada caso evaluamos las siguientes variables: indicación de la ecocardiografía fetal, EG al diagnóstico, presencia o no de anomalías extracardíacas, cariotipo, técnica de obtención del cariotipo y tipo de anomalía cromosómica. Analizamos también la concordancia diagnóstica pre-posnatal y la evolución de los pacientes hasta el período neonatal.
Estudio estadísticoSe realizó un estudio descriptivo de las distintas variables analizadas. La distribución de las variables cualitativas se representó según su frecuencia absoluta y porcentaje. Las variables cuantitativas se describieron mediante su media acompañada de la desviación estándar. El estudio de asociación entre variables cualitativas se realizó mediante la prueba de la chi-cuadrado o la prueba exacta de Fisher. La cuantificación de la magnitud del efecto fue valorada a través del riesgo relativo junto con su intervalo de confianza al 95%. Para el análisis de los datos se utilizó el programa SPSS, versión 17.0 (SPSS, Chicago, IL, EE. UU.).
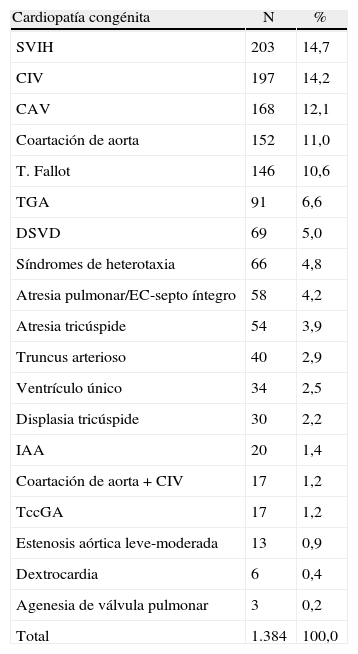
ResultadosEn el período de estudio se diagnosticaron un total de 1.425 CC pero 41 fueron excluidas por no disponer de estudio cromosómico (n=6) o por no disponer de comprobación posnatal de la existencia de CC (n=35), y estas por negativa de los padres a la necropsia (n=4), por tener lugar la IVE en otra institución y no ser seguida de necropsia (n=27), por la intensa maceración fetal tras la muerte intrauterina en gestación gemelar (n=2), o por pérdida en el seguimiento (n=2). Por tanto, la población de estudio final estuvo constituida por 1.384 fetos. En la tabla 1 se muestra la distribución de las CC diagnosticadas prenatalmente.
Distribución de las cardiopatías congénitas diagnosticadas prenatalmente en el período de estudio ordenadas por orden de frecuencia
| Cardiopatía congénita | N | % |
| SVIH | 203 | 14,7 |
| CIV | 197 | 14,2 |
| CAV | 168 | 12,1 |
| Coartación de aorta | 152 | 11,0 |
| T. Fallot | 146 | 10,6 |
| TGA | 91 | 6,6 |
| DSVD | 69 | 5,0 |
| Síndromes de heterotaxia | 66 | 4,8 |
| Atresia pulmonar/EC-septo íntegro | 58 | 4,2 |
| Atresia tricúspide | 54 | 3,9 |
| Truncus arterioso | 40 | 2,9 |
| Ventrículo único | 34 | 2,5 |
| Displasia tricúspide | 30 | 2,2 |
| IAA | 20 | 1,4 |
| Coartación de aorta + CIV | 17 | 1,2 |
| TccGA | 17 | 1,2 |
| Estenosis aórtica leve-moderada | 13 | 0,9 |
| Dextrocardia | 6 | 0,4 |
| Agenesia de válvula pulmonar | 3 | 0,2 |
| Total | 1.384 | 100,0 |
CAV: canal aurículo-ventricular; CIV: comunicación interventricular; DSVD: doble salida del ventrículo derecho; EC: estenosis crítica; IAA: interrupción del arco aórtico; SVIH: síndrome de ventrículo izquierdo hipoplásico, incluyendo formas de estenosis aórtica crítica; T. Fallot: tetralogía de Fallot, incluyendo las formas con atresia o agenesia de la válvula pulmonar; TccGA: transposición congénitamente corregida de grandes arterias; TGA: transposición de grandes arterias.
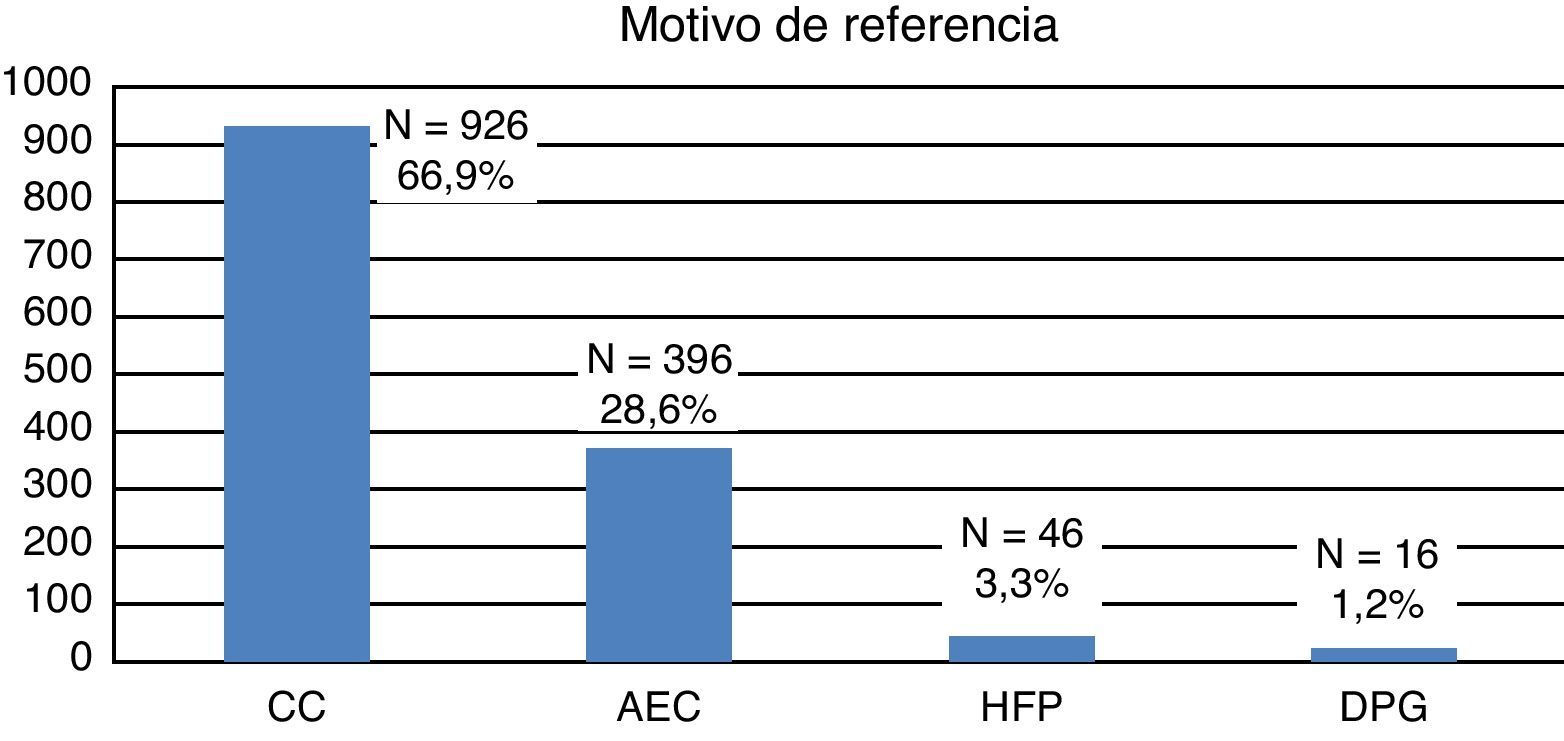
La mayoría de las gestantes (924; 66,8%) procedían de otras áreas sanitarias y nos fueron remitidas para ecocardiografía, y eventual asesoramiento y manejo perinatal. El resto (460; 33,2%) pertenecían a nuestro centro, donde tenía lugar tanto la vigilancia prenatal habitual como las ecografías obstétricas. Como muestra la figura 1, el motivo de referencia más frecuente para la realización de una ecocardiografía fetal fue la sospecha de CC en una ecografía obstétrica rutinaria (66%). Unicamente en el 3,3% había antecedentes familiares de CC.

En 919 casos (66,4%), la CC se presentaba de forma aislada y en el resto (n=465; 33,6%) existían otras anomalías asociadas, de tipo malformativo o estigmas de cromosomopatía. Se dispone de información relativa a la TN en 988 casos y en 215 (21,8%) estaba >p95.
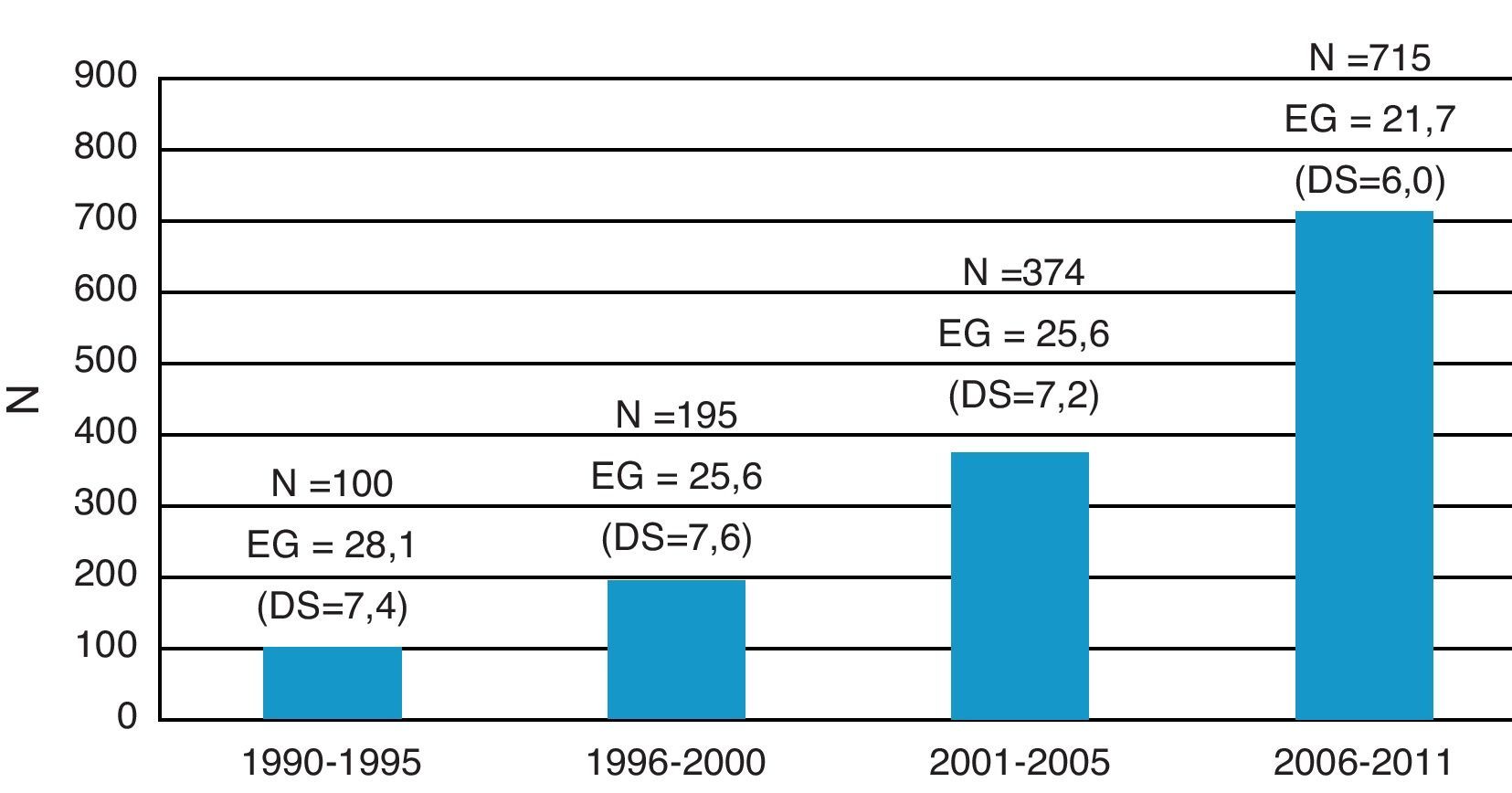
La EG media al diagnóstico fue de 23,1 semanas (DS:6,28). Esta disminuyó a lo largo del período de estudio de modo que entre 1990 y 1995 fue de 28 semanas (DS:7,4) y entre 2006 y 2011 fue de 21,6 semanas (DS:6,0) (p<0,001). En 813 casos (58,7%) el diagnóstico se realizó ≤ 22 semanas de gestación. La distribución por quinquenios de las CC incluidas en el estudio y su edad gestacional al diagnóstico se muestran en la figura 2.

Se realizó estudio prenatal del cariotipo en la mayoría de los casos (n=848; 61,3%) mediante amniocentesis (n=486), cordocentesis (n=268) o biopsia corial (n=92). En el resto de los casos (n=536) bien se estudió el cariotipo posnatalmente (172; 12,4%) o bien se excluyó clínicamente la presencia de cromosomopatía dada la ausencia de otras anomalías asociadas a la CC y de marcadores clínicos indicativos de aquella (364; 26,3%). El 26,2% de los fetos diagnosticados de CC tenían una anomalía cromosómica asociada (363/1.384). En la tabla 2 se muestra la distribución de las cromosomopatías diagnosticadas. En la mayoría de los casos el diagnóstico de la cromosomopatía fue prenatal (324/363; 89,3%) y solo en 39 casos (10,7%) fue posnatal. En estos casos, la indicación para no realizar el estudio prenatal del cariotipo fue la negativa de los padres (n=28), el deterioro fetal avanzado que recomendaba la finalización inmediata de la gestación (n=3) y la avanzada EG al diagnóstico (≥37 semanas) (n=7). La EG al diagnóstico fue menor en las CC asociadas a anomalías cromosómicas (19,6 semanas, DS 7,2) frente a la de las CC sin cromosomopatía (24,2 semanas, DS 6,5) (p < 0,001).
Distribución de las cromosomopatías diagnosticadas prenatalmente en los fetos con cardiopatías congénitas durante el período de estudio, ordenadas por orden de frecuencia
| Cromosomopatía | N | % |
| Trisomía 21 | 119 | 32,8 |
| Trisomía 18 | 107 | 29,5 |
| Trisomía 13 | 40 | 11,0 |
| Monosomía X0 | 34 | 9,4 |
| Microdeleción 22q11* | 25 | 6,9* |
| Otras | 38 | 10,5 |
| Triploidía | 11 | |
| 47XXY | 1 | |
| 47XXX | 2 | |
| Derivados de traslocación | 9 | |
| 46XX, der(9) t(9;16)(p12;?) de novo | ||
| 46XY, der(11)t(7;11)(q11;q25) pat | ||
| 46XX, der(15)dup(15)(q15,q25) de novo | ||
| 46XX, der(15)t(2,15) pat | ||
| 46XY, der(5) t(5;?)(5q23;?) de novo | ||
| 46XX, der(4) dup(4)(q21,qter) de novo | ||
| 46XX, der(22) t(22;?)(q13;?) de novo | ||
| 46XY, der(6)t(6;13) pat | ||
| 46XY, der(6)t(6;15) mat | ||
| Deleciones | 3 | |
| 46XX, del(13)(q31,qter) de novo | ||
| 46XX, del(16)(q22,q23) de novo | ||
| 46XX, del(8)(p21,pter) de novo | ||
| Traslocaciones recíprocas | 2 | |
| 46XY,t(15;16) de novo | ||
| 46XX,t(2;15) de novo | ||
| Cromosomas en anillo | 1 | |
| 46XY/46XY,r(4)/45XY,-4 de novo | ||
| Cromosomas marcadores | 3 | |
| 47XY,+marc i(12p) de novo | ||
| 47XX,+marc idic(22) de novo | ||
| 47XY,+marc, i(15p) de novo | ||
| Inversiones | ||
| 46XX, inv(1)(p21,q43) de novo | 1 | |
| Trisomía 7 | 1 | |
| Trisomía 9 | 1 | |
| Trisomía 2 | 1 | |
| Doble aneuploidía 46X0, +21 | 1 | |
| Tetraploidía 92XXYY | 1 | |
| Total | 363 | |
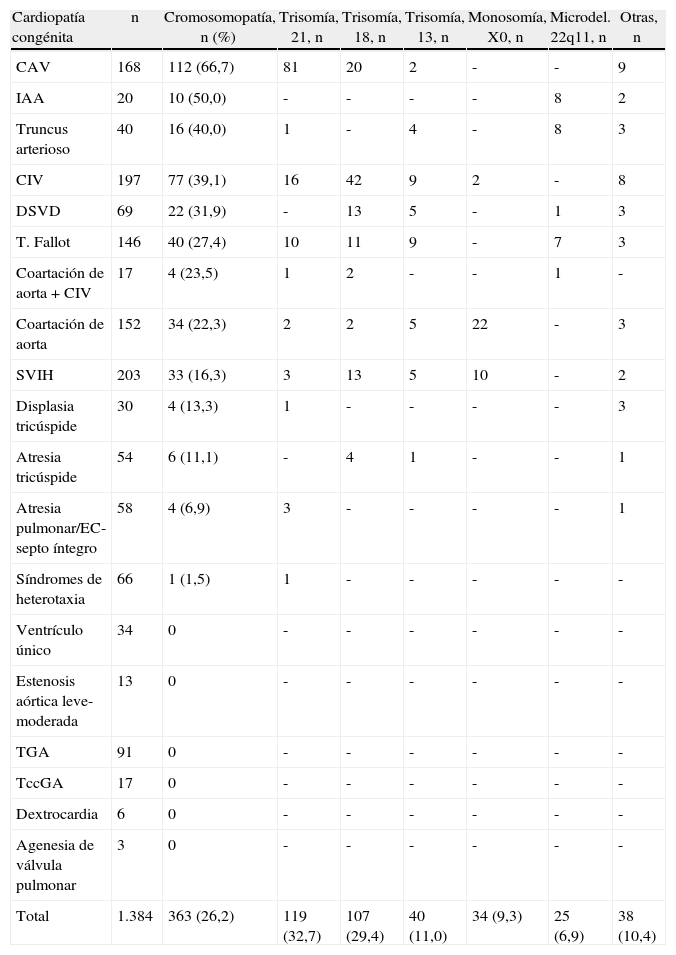
En la tabla 3 se muestra la asociación entre las diferentes CC y las anomalías cromosómicas. Las CC que mostraron una mayor asociación con cromosomopatías fueron el canal aurículo-ventricular ([CAV], 66,7%), la interrupción del arco aórtico ([IAA], 50%) y el truncus arterioso (40%). En este sentido, aunque se observó una mayor relación entre ciertas CC y algunas anomalías cromosómicas, como por ejemplo entre el CAV y la trisomía 21 (48,2%), la IAA y el truncus arterioso con la microdeleción 22q11 (40 y 20%, respectivamente), la comunicación interventricular (CIV) y la doble salida del ventrículo derecho (DSVD) con la trisomía 18 (21,3 y 18,8%, respectivamente) y la coartación de aorta con la monosomía X0 (14,5%), esta relación en ningún caso fue de exclusividad. Por el contrario, no se observaron anomalías cromosómicas en CC tales como la TGA, TccGA, VU, agenesia de válvula pulmonar, formas leves-moderadas de estenosis de la válvula aórtica y dextrocardia.
Asociación con cromosomopatías y distribución de las mismas en las diferentes cardiopatías congénitas incluidas en el estudio, ordenadas según la frecuencia de asociación
| Cardiopatía congénita | n | Cromosomopatía, n (%) | Trisomía, 21, n | Trisomía, 18, n | Trisomía, 13, n | Monosomía, X0, n | Microdel. 22q11, n | Otras, n |
| CAV | 168 | 112 (66,7) | 81 | 20 | 2 | - | - | 9 |
| IAA | 20 | 10 (50,0) | - | - | - | - | 8 | 2 |
| Truncus arterioso | 40 | 16 (40,0) | 1 | - | 4 | - | 8 | 3 |
| CIV | 197 | 77 (39,1) | 16 | 42 | 9 | 2 | - | 8 |
| DSVD | 69 | 22 (31,9) | - | 13 | 5 | - | 1 | 3 |
| T. Fallot | 146 | 40 (27,4) | 10 | 11 | 9 | - | 7 | 3 |
| Coartación de aorta + CIV | 17 | 4 (23,5) | 1 | 2 | - | - | 1 | - |
| Coartación de aorta | 152 | 34 (22,3) | 2 | 2 | 5 | 22 | - | 3 |
| SVIH | 203 | 33 (16,3) | 3 | 13 | 5 | 10 | - | 2 |
| Displasia tricúspide | 30 | 4 (13,3) | 1 | - | - | - | - | 3 |
| Atresia tricúspide | 54 | 6 (11,1) | - | 4 | 1 | - | - | 1 |
| Atresia pulmonar/EC-septo íntegro | 58 | 4 (6,9) | 3 | - | - | - | - | 1 |
| Síndromes de heterotaxia | 66 | 1 (1,5) | 1 | - | - | - | - | - |
| Ventrículo único | 34 | 0 | - | - | - | - | - | - |
| Estenosis aórtica leve-moderada | 13 | 0 | - | - | - | - | - | - |
| TGA | 91 | 0 | - | - | - | - | - | - |
| TccGA | 17 | 0 | - | - | - | - | - | - |
| Dextrocardia | 6 | 0 | - | - | - | - | - | - |
| Agenesia de válvula pulmonar | 3 | 0 | - | - | - | - | - | - |
| Total | 1.384 | 363 (26,2) | 119 (32,7) | 107 (29,4) | 40 (11,0) | 34 (9,3) | 25 (6,9) | 38 (10,4) |
CAV: canal aurículo-ventricular; CIV: comunicación interventricular; DSVD: doble salida del ventrículo derecho; EC: estenosis crítica; IAA: interrupción del arco aórtico; SVIH: síndrome de ventrículo izquierdo hipoplásico, incluyendo formas de estenosis aórtica crítica; T. Fallot: tetralogía de Fallot, incluyendo las formas con atresia o agenesia de la válvula pulmonar; TccGA: transposición congénitamente corregida de grandes arterias; TGA: transposición de grandes arterias.
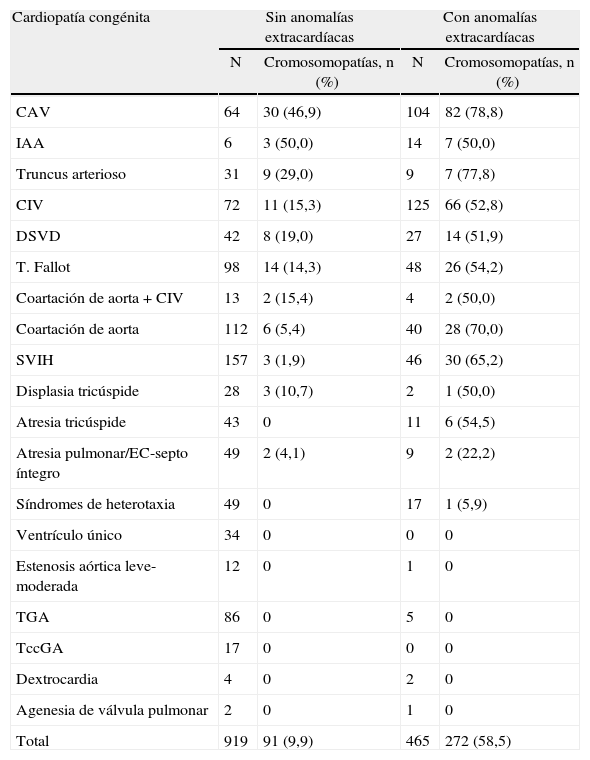
En la tabla 4 se muestra la asociación de las CC con cromosomopatías en función de la existencia o no de otras anomalías asociadas. La frecuencia observada de anomalías cromosómicas en las CC aisladas fue del 9,9% (91/919) mientras que en aquellos casos en que coexistían otras anomalías extracardíacas esta tasa fue del 58,5% (272/465). Por tanto, el riesgo de tener una anomalía cromosómica es significativamente mayor en los fetos cuya CC se asocia a otras anomalías extracardíacas frente a aquellos en que la CC se presenta de forma aislada (RR=6,0, IC 95% 4,8-7,4; p<0,001). Concretamente, en 113 de los 215 fetos con CC que tenían o habían tenido una TN aumentada se diagnosticó una cromosomopatía (52,6%). Para todas las CC la tasa de cromosomopatías aumentó cuando existían otras anomalías extracardíacas, con la excepción de la IAA en que dicha tasa se mantuvo similar respecto a los casos de IAA «aislada». De hecho, en los casos de atresia tricúspide aislada o de síndrome de heterotaxia en que no había ninguna anomalía ajena a las que pueden formar parte del propio síndrome, no observamos ninguna cromosomopatía y, del mismo modo, la tasa de cromosomopatías fue muy baja en los casos de ventrículo izquierdo hipoplásico aislado.
Asociación con cromosomopatías de las diferentes cardiopatías congénitas incluidas en el estudio en función de la presencia o ausencia de otras anomalías extracardíacas
| Cardiopatía congénita | Sin anomalías extracardíacas | Con anomalías extracardíacas | ||
| N | Cromosomopatías, n (%) | N | Cromosomopatías, n (%) | |
| CAV | 64 | 30 (46,9) | 104 | 82 (78,8) |
| IAA | 6 | 3 (50,0) | 14 | 7 (50,0) |
| Truncus arterioso | 31 | 9 (29,0) | 9 | 7 (77,8) |
| CIV | 72 | 11 (15,3) | 125 | 66 (52,8) |
| DSVD | 42 | 8 (19,0) | 27 | 14 (51,9) |
| T. Fallot | 98 | 14 (14,3) | 48 | 26 (54,2) |
| Coartación de aorta + CIV | 13 | 2 (15,4) | 4 | 2 (50,0) |
| Coartación de aorta | 112 | 6 (5,4) | 40 | 28 (70,0) |
| SVIH | 157 | 3 (1,9) | 46 | 30 (65,2) |
| Displasia tricúspide | 28 | 3 (10,7) | 2 | 1 (50,0) |
| Atresia tricúspide | 43 | 0 | 11 | 6 (54,5) |
| Atresia pulmonar/EC-septo íntegro | 49 | 2 (4,1) | 9 | 2 (22,2) |
| Síndromes de heterotaxia | 49 | 0 | 17 | 1 (5,9) |
| Ventrículo único | 34 | 0 | 0 | 0 |
| Estenosis aórtica leve-moderada | 12 | 0 | 1 | 0 |
| TGA | 86 | 0 | 5 | 0 |
| TccGA | 17 | 0 | 0 | 0 |
| Dextrocardia | 4 | 0 | 2 | 0 |
| Agenesia de válvula pulmonar | 2 | 0 | 1 | 0 |
| Total | 919 | 91 (9,9) | 465 | 272 (58,5) |
CAV: canal aurículo-ventricular; CIV: comunicación interventricular; DSVD: doble salida del ventrículo derecho; EC: estenosis crítica; IAA: interrupción del arco aórtico; SVIH: síndrome de ventrículo izquierdo hipoplásico, incluyendo formas de estenosis aórtica crítica; T. Fallot: tetralogía de Fallot, incluyendo las formas con atresia o agenesia de la válvula pulmonar; TccGA: transposición congénitamente corregida de grandes arterias; TGA: transposición de grandes arterias.
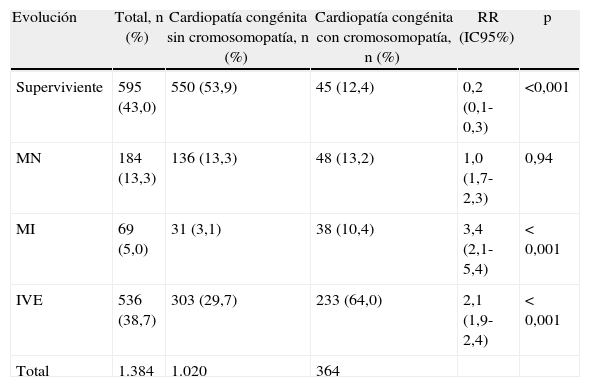
En la tabla 5 se muestra el resultado de la gestación tanto del grupo de estudio global como en función de la existencia o no de cromosomopatía. Observamos diferencias significativas tanto en la tasa de supervivientes al período neonatal como de mortalidad intrauterina y de IVE entre los fetos con cromosomopatía y los que tenían cariotipo normal. En la inmensa mayoría de los casos, el diagnóstico prenatal de la CC fue confirmado posnatalmente (1.319/1.384; 95,3%), bien total (n=1.185) o parcialmente (n=134). Hubo 65 falsos positivos (4,7%) que correspondieron a sospechas de coartación de aorta que no fueron confirmadas posnatalmente (n=47), defectos septales que no se observaron al nacimiento o en la necropsia (n=16) y estenosis valvulares no confirmadas (n=2).
Resultado de la gestación tanto en el grupo de estudio global como en función de la existencia o no de cromosomopatía
| Evolución | Total, n (%) | Cardiopatía congénita sin cromosomopatía, n (%) | Cardiopatía congénita con cromosomopatía, n (%) | RR (IC95%) | p |
| Superviviente | 595 (43,0) | 550 (53,9) | 45 (12,4) | 0,2 (0,1-0,3) | <0,001 |
| MN | 184 (13,3) | 136 (13,3) | 48 (13,2) | 1,0 (1,7-2,3) | 0,94 |
| MI | 69 (5,0) | 31 (3,1) | 38 (10,4) | 3,4 (2,1-5,4) | <0,001 |
| IVE | 536 (38,7) | 303 (29,7) | 233 (64,0) | 2,1 (1,9-2,4) | <0,001 |
| Total | 1.384 | 1.020 | 364 |
IVE: interrupción voluntaria del embarazo; MI: muerte intrauterina; MN: muerte neonatal.
Este estudio confirma que la prevalencia de anomalías cromosómicas en los fetos con CC es alta, y la tasa observada (26,2%) se correlaciona bien con la reportada en otras series fetales, que oscila entre el 11,3 y el 29,3%9,11,14–16. Esta variabilidad puede ser atribuida, en gran medida, a la diferente distribución de las CC en cada serie, dada la muy diferente relación con las cromosomopatías de las distintas CC11,15–17. En efecto, mientras que la probabilidad de cromosomopatía es muy elevada en CC tales como el CAV, el truncus arterioso, la IAA o la DSVD, en otras como la TGA, la TccGA, el VU o las formas leves-moderadas de estenosis aórtica, esta relación es nula.
Por otra parte, la asociación de las CC con cromosomopatías es claramente dependiente de la coexistencia con otras anomalías anatómicas extracardíacas de modo que en estos casos el riesgo de cromosomopatía es 6 veces superior. Este aumento del riesgo afecta prácticamente a todas las CC analizadas en este estudio. Siendo cierto que las anomalías extracardíacas, cuando están presentes, tienen un efecto multiplicador del riesgo de cromosomopatía, también es conveniente destacar que cuando están ausentes se reduce la probabilidad de cromosomopatía hasta el punto de que en algunos casos como la atresia tricúspide aislada o los síndromes de heterotaxia la relación con cromosomopatías se anula. Por ello, son desafíos esenciales del diagnóstico prenatal de las CC la precisión diagnóstica y el análisis morfológico extracardíaco exhaustivo, incluyendo la revisión de la información recogida en exploraciones previas, dada la dependencia que de estos factores tiene la probabilidad de que el feto tenga una cromosomopatía y, por tanto, el impacto que puede tener en nuestra recomendación, más o menos contundente, de someter el embarazo a una técnica invasiva. Aunque la decisión final de realizar o no una técnica invasiva para diagnóstico prenatal es competencia siempre de los padres, a la vista de los resultados de nuestra serie, una de las mayores publicadas hasta la fecha proveniente de un único centro, y a que están en consonancia con los descritos en otras series9,15–20, consideramos que nuestra recomendación de someter el embarazo a dicha prueba tras el diagnóstico prenatal de una CC debe dejar de ser monótona y sistemática y debe pasar a ser una recomendación individualizada, CC-dependiente y anomalías extracardíacas-dependiente, de manera que en determinadas CC y más aún si se presentan aisladas, podemos apoyar su no realización9,10,16. Esta conducta permitiría evitar la realización de un 15-20% de técnicas invasivas en fetos con CC. Esto es especialmente importante si tenemos en cuenta que estas técnicas no están exentas de riesgo21. No obstante, y como muestran nuestros resultados, en los casos indicados el estudio del cariotipo resulta esencial dada su importancia en la decisión final de los padres sobre la continuación o no del embarazo y su impacto en la supervivencia.
De acuerdo con otras series15,17, hemos observado que aunque en las CC asociadas a anomalías cromosómicas parece existir una relación preferencial de determinadas CC con algunas cromosomopatías, realmente puede presentarse cualquiera de estas. Estos resultados apoyan la hipótesis de que muchos genes situados en diferentes cromosomas están implicados en la relación de causalidad de las CC7.
La distribución de las diferentes CC en nuestra serie coincide con otras series fetales 17,22–24, observándose cómo los defectos septales, la patología obstructiva del corazón izquierdo y el grupo de las CC conotruncales representan el 80% de las CC diagnosticadas prenatalmente. También es similar la tasa de anomalías asociadas. Todo esto enfatiza la importancia de, por una parte, explorar los tractos de salida y, por otra, hacer un examen anatómico fetal minucioso cada vez que se diagnostique una CC. Por lo demás, y como era de esperar, hemos asistido a lo largo del tiempo a un aumento progresivo en el número de CC diagnosticadas prenatalmente y a una disminución de la EG al diagnóstico, producto tanto de la mejora en la exploración ecográfica obstétrica de cribado, que sigue siendo la indicación de ecocardiografía que más diagnósticos prenatales de CC permite, como de la incorporación de nuevas indicaciones de ecocardiografía fetal, como la TN aumentada25,26. Por su parte, confirmamos que la ecocardiografía fetal tiene un elevado grado de precisión diagnóstica6,27-29 acumulándose los falsos positivos en las sospechas de coartación de aorta, como consecuencia de que su diagnóstico habitualmente reside en signos indirectos y no específicos30, y en los defectos del septo interventricular, aunque estos pueden cerrar intraútero de manera espontánea31.
Destacaríamos 2 limitaciones principales en nuestra serie. En primer lugar, que en un elevado número de casos no disponemos del resultado del estudio citogenético, aunque realmente consideramos altamente improbable que se hayan dejado de diagnosticar anomalías cromosómicas relevantes al no haber indicios clínicos de la existencia de cromosomopatía más allá de la propia CC en estos pacientes. En segundo lugar, es posible que nuestros resultados subestimen la contribución que tienen en la etiología de las CC las alteraciones genéticas que no se pueden detectar mediante la utilización de técnicas de citogenética convencional, dado que la determinación de la microdeleción 22q11 no ha estado disponible durante todo el período de estudio. No obstante, la incorporación de herramientas de citogenómica como el multiplex ligation-dependent probe amplification (MLPA) permiten ampliar el horizonte diagnóstico de la FISH poniendo el punto de partida para el desarrollo de técnicas de array-CGH y de secuenciación masiva que harán posible la identificación de nuevas regiones cromosómicas asociadas a CC, fundamentalmente en formas sindrómicas, así como el diagnóstico de algunos casos de formas no sindrómicas32,33.
En conclusión, con la solidez de los números aportados en esta serie, consideramos que aún siendo enormemente relevante la información del cariotipo en los fetos con CC para la toma de decisiones de los padres y el pronóstico del paciente, la recomendación de realizar dicho estudio ha de individualizarse según las características de cada caso, pudiendo evitarse los riesgos de la técnica invasiva diagnóstica en un notable porcentaje de casos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.