


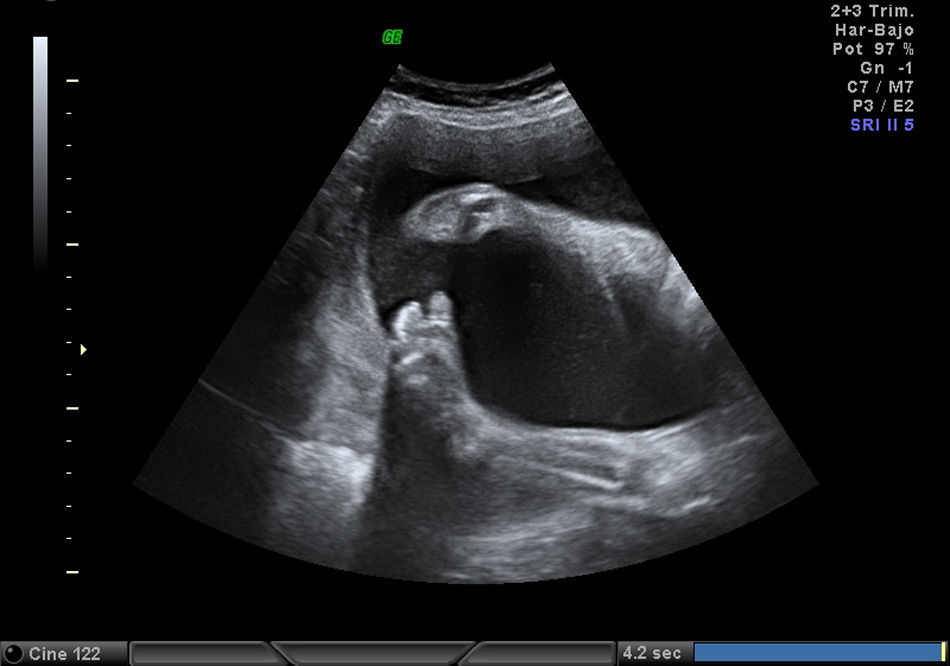
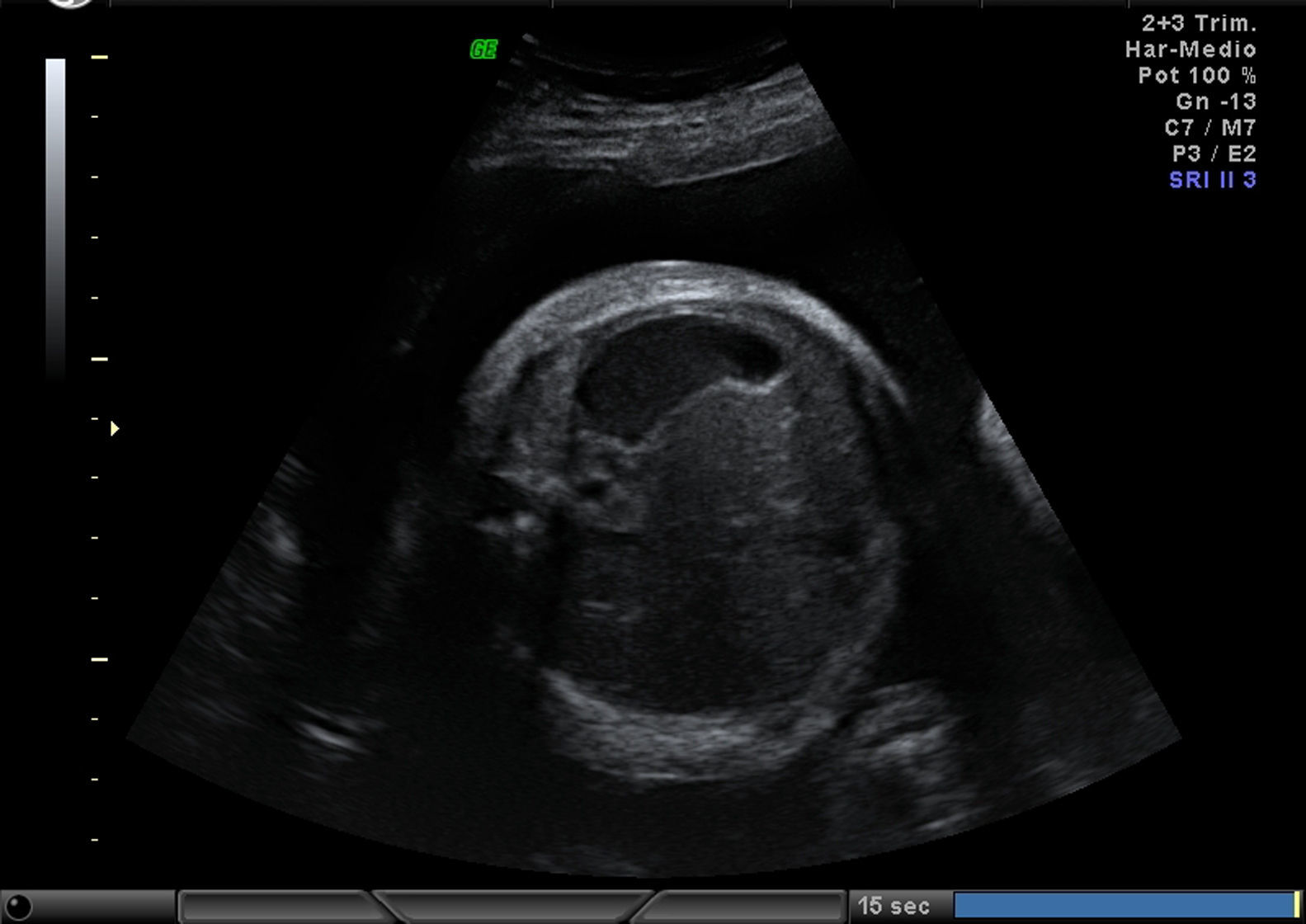
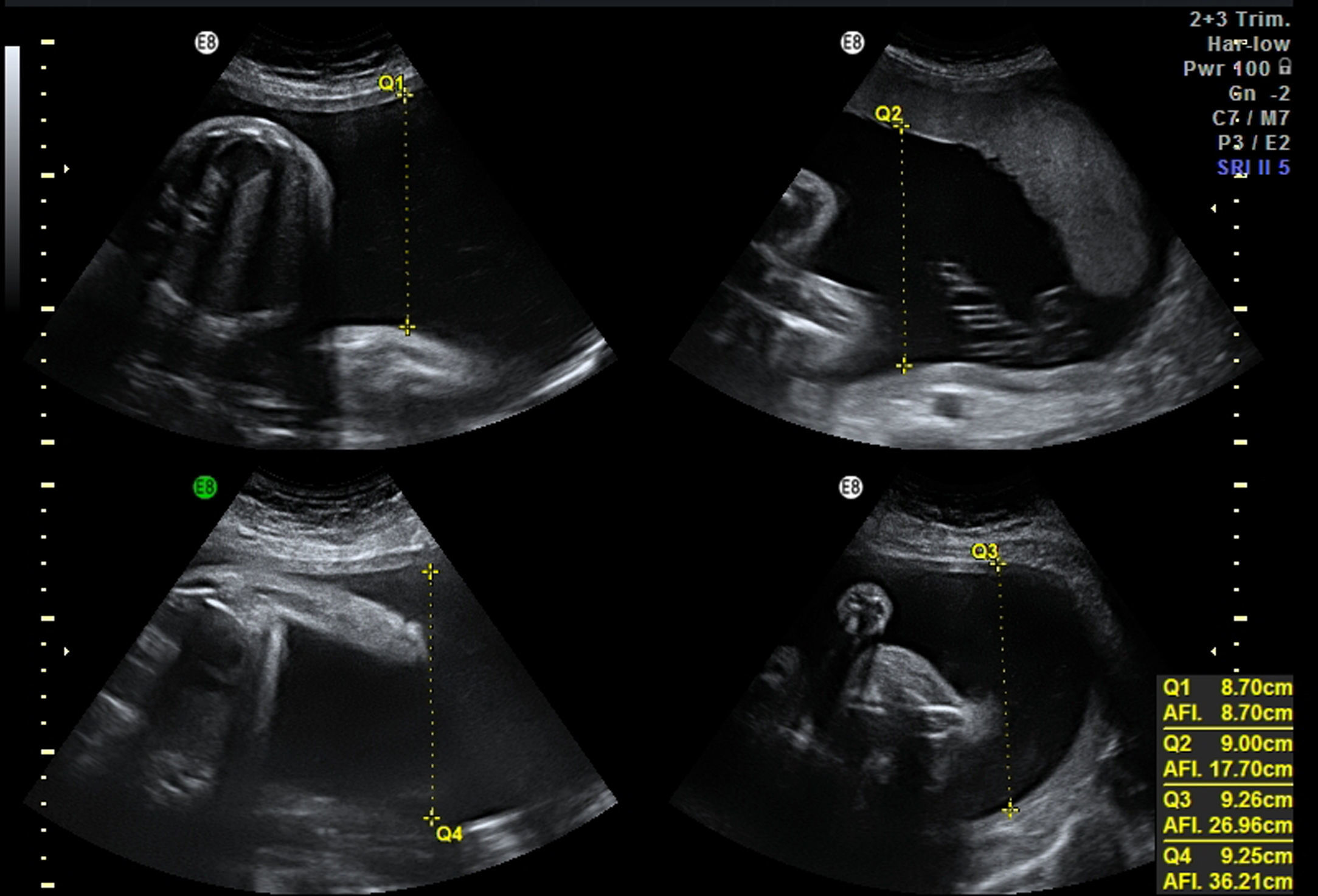
Mujer de 34 años, sin antecedentes obstétricos de interés. Gestante mediante fecundación in vitro por factor masculino. Cribado bioquímico de riesgo bajo 1/1.016. Marcadores ecográficos: translucencia nucal de 1,5, ductus venoso normal. Amniocentesis por ansiedad: 46,XX. Ecografía morfológica de 20 semanas normal. En un seguimiento a las 28 semanas se detecta anomalía posicional severa de ambos pies (figs. 1 and 2) y otros hallazgos evidentes en las imágenes siguientes (figs. 3–6).






Se revisa la iconografía del control ecográfico de las 20 semanas y se confirma la ausencia de anomalías en ambos pies en esa exploración, esto orienta como posible causa una afección evolutiva.
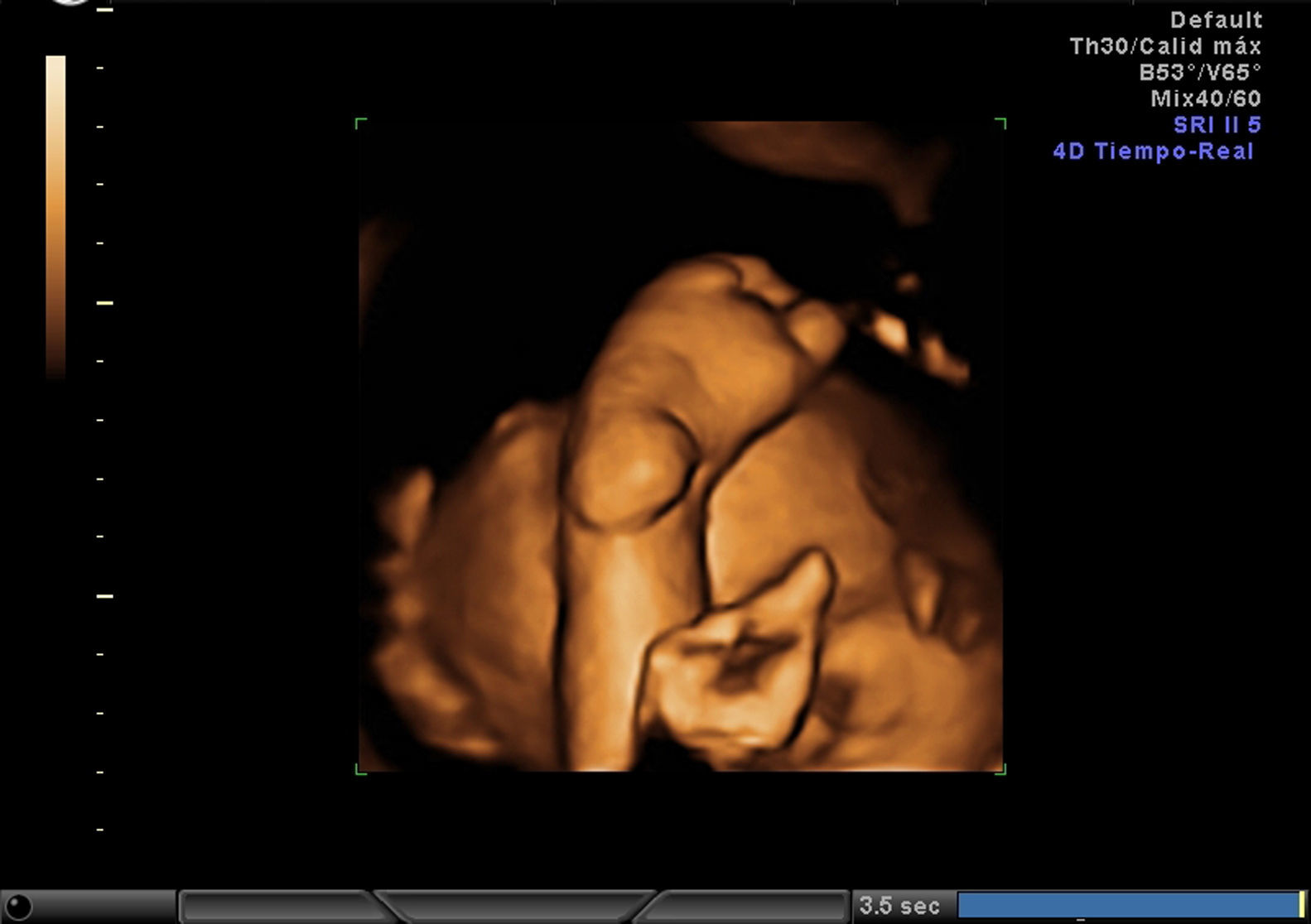
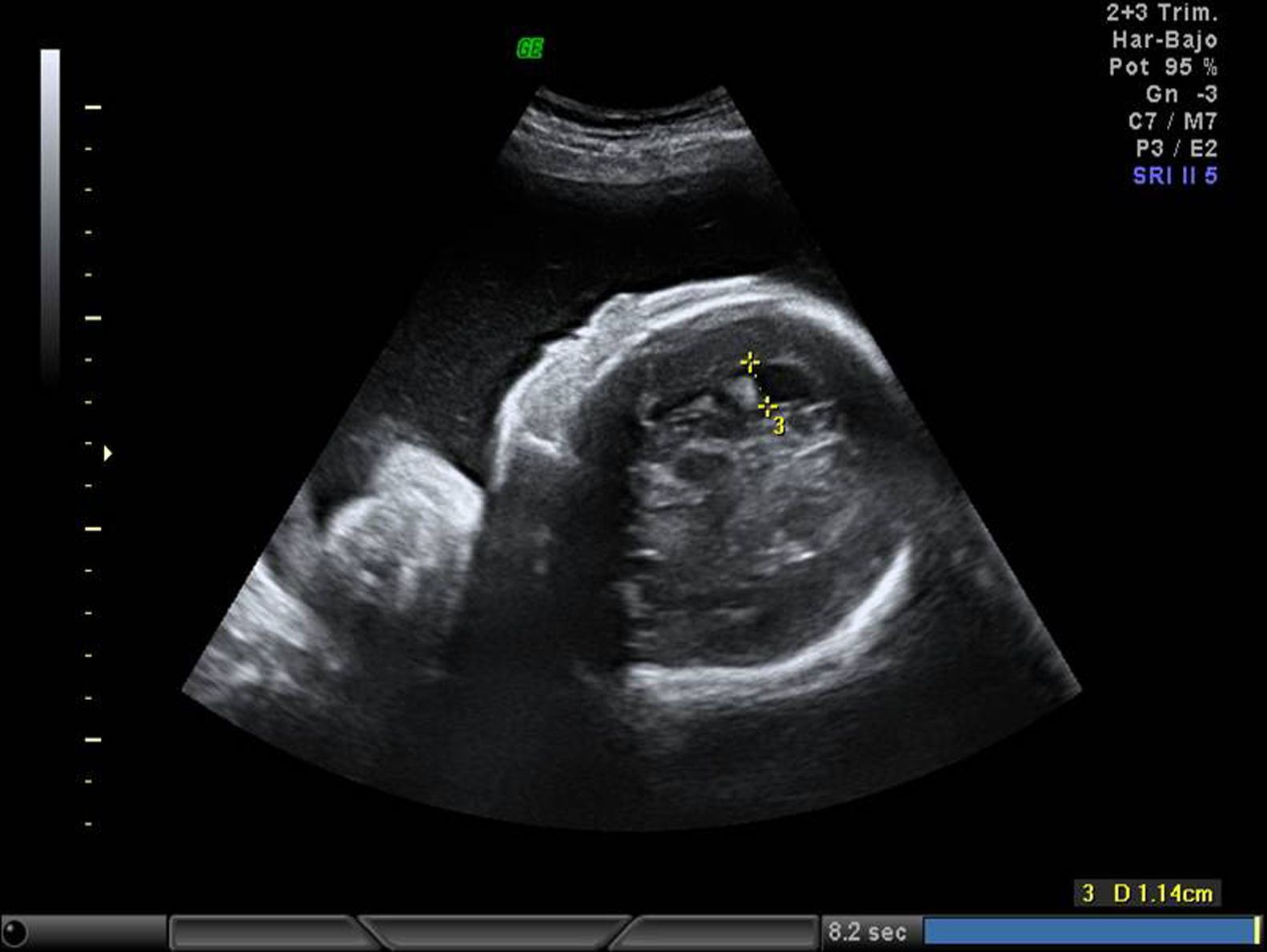
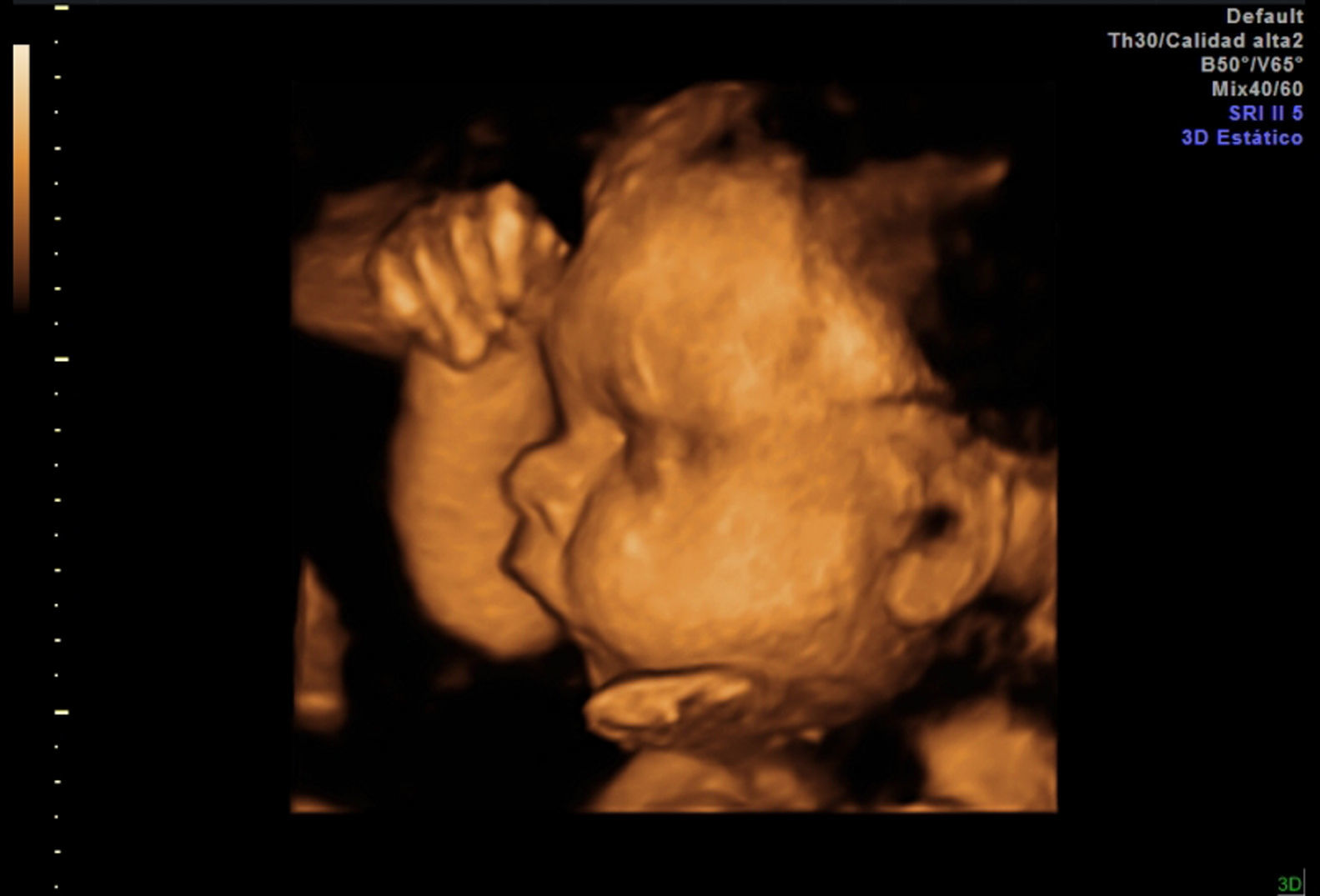
EvoluciónLa detección prenatal de anomalía posicional de los pies (figs. 1 and 2) asociada a otras anomalías, como ventriculomegalia (fig. 3), retrognatia (fig. 4), polihidramnios (índice de líquido amniótico de 38cm) (fig. 6), es indicativa de síndrome. En el momento del diagnóstico (28 semanas), como consecuencia del polihidramnios, la longitud cervical era de 10mm y a pesar del tratamiento con indometacina se produjo parto prematuro a las 33 semanas, con un peso de 1.990g y Apgar: 3 al minuto. El recién nacido precisó intubación a los 5 minutos de vida e ingreso en la unidad de cuidados intensivos neonatal. A la exploración presentaba hipotonía generalizada, arreflexia, megacefalia (fontanela normotensa) y braquicefalia. La ecografía cerebral indicaba ventriculomegalia moderada. El diagnóstico probable era de distrofia muscular. En el estudio materno se observó electromiograma con numerosos fenómenos miotónicos, compatibles con enfermedad de Steinert. El estudio molecular en la madre y en el recién nacido indicó positivo para distrofia miotónica de Steinert. Debido al mal pronóstico de la distrofia miotónica de Steinert congénita, las maniobras terapéuticas se limitan hasta producirse fallecimiento neonatal.
DiagnósticoDistrofia miotónica de Steinert forma congénita, de herencia autonómica dominante con madre afectada no diagnosticada.
ComentariosLa enfermedad de Steinert es la distrofia muscular más frecuente en adultos, y tiene una gran variedad de expresiones clínicas. En este caso clínico la madre estaba afectada, pero no diagnosticada, porque la clínica era prácticamente nula. A partir de la sospecha clínica en el recién nacido, se diagnostica a la familia (tres generaciones estudiadas afectadas). La forma congénita de la enfermedad de Steinert es una de las causas más frecuentes de hipotonía neonatal, es grave y de mal pronóstico. El diagnóstico molecular corresponde a una mutación en la región q13.3 del cromosoma 19 con repetición del nucleótido CTG en región no codificante de un gen de proteinacinasa. El número de copias de tripletes de CTG está relacionado con la gravedad del cuadro clínico. En la forma clásica hay unas 200 repeticiones y en la forma congénita pueden haber hasta 2.000 repeticiones (el número de repeticiones aumenta con el número de generaciones afectadas).
Resolución de caso iconográfico presentado en el número 3/2009.