



Presentamos un caso de trisomía del cromosoma 8 mosaico, diagnosticado mediante citogenética con técnicas de hibridación in situ con fluorescencia (FISH) tras amniocentesis a las 18 semanas de la gestación, en una paciente que en el estudio ecográfico mostró imágenes de quiste unilateral de los plexos coroideos, así como un hemangioma placentario.
Se hace una valoración de los hallazgos fenotípicos fetales en el estudio necrópsico, que se comparan con los descritos en diferentes casos de trisomías totales y/o parciales del cromosoma 8, y se valora muy especialmente la relación existente entre la presencia de quistes de los plexos coroideos y la aparición de cromosomopatías.
We report a case of trisomy 8 mosaicism, diagnosed by FISH (fluorescent in situ hibridization) after 18 week pregnancy amniocentesis, in a patient whose ultrasound scan showed a unilateral choroid plexus cyst and placental haemangioma.
We assessed the foetopathological examination, comparing it with different cases of total and partial trisomies of chromosome 8, paying special attention to the relationship between choroid plexus cyst and chromosomopathies.
La trisomía 8, también conocida como síndrome de Warkany, es una enfermedad que aparece en el 0,8% de los abortos espontáneos y se estima que ocurre en el 0,1% de todos los embarazos clínicamente reconocibles1.
Los pacientes con trisomía del grupo C se han diagnosticado desde 1963, pero la trisomía 8 mosaico la describieron por primera vez Grouchy et al2 en 1971 y el síndrome lo describió Rethore3 en 1977. En recién nacidos vivos, su frecuencia se estima en 1:25.000-50.000 con una predilección 5:1 por el sexo masculino4 y asociada casi siempre a un mosaicismo. La línea celular anormal tiende a desaparecer de los linfocitos con la edad, y así, en pacientes adultos la aneuploidía muchas veces sólo se puede demostrar en cultivo de fibroblastos5,6.
Los pacientes con trisomía 8 mosaico se caracterizan por la presencia de una dismorfia facial muy indicativa, así como anomalías osteoarticulares, algunas bastante graves. Pueden mostrar una gran variedad de imágenes fenotípicas, incluido:
- a)
Dismorfia craneofacial: macrocefalia con frente alta y prominente, cara alargada, moderado hipertelorismo con presencia ocasional de ptosis y estrabismo. Nariz ancha y achatada. Labio superior grande, pero es el labio inferior el que resulta especialmente significativo por ser grueso y evertido. Retromicrognatia. Orejas de forma anormal con aurícula larga y antihélix prominente.
- b)
Cuello, tórax y abdomen: cuello corto y ancho que contrasta con los hombros estrechos y el tronco largo. Suelen existir malformaciones esqueléticas, incluido cifoescoliosis dorso-lumbar, vértebras anormales o supernumerarias, espina bífida, costillas supernumerarias, pecho en embudo y pelvis estrecha o hipoplásica.
- c)
Extremidades: con frecuencia hay lesiones osteoarticulares que incluyen braquidactilia o aracnodactilia, clinodactilia, campodactilia y ausencia bilateral de rótula. Las anomalías articulares no son detectables en el momento del nacimiento, pero con la edad aparecen contracturas que provocan deformidades. En niños jóvenes aparece un síntoma bastante característico: surcos palmares y plantares profundos7. Las uñas son hipoplásicas y convexas y pueden estar ausentes al nacer.
- d)
Genitales: en niños se ha descrito criptorquidia, hipoplasia testicular y pubertad retrasada. Se observan pacientes adultos con fenotipo bastante normal que consultan por esterilidad primaria por oligospermia grave; sin embargo, no hay datos precisos sobre fertilidad en pacientes con trisomía 88.
- e)
Malformaciones: las malformaciones internas son raras y sin peligro para la vida del paciente. Se pueden observar malformaciones cardíacas, pero las renales y ureterales son las malformaciones que podrían considerarse más características. Se ha descrito la existencia de agencia del cuerpo calloso9,10.
- f)
Retraso mental: no suele ser muy llamativo. El IQ varía entre 12 y 94, con una media entre 45 y 75. De hecho, la mayoría de los casos se diagnostican en adultos sin grandes alteraciones del fenotipo11. Se han descrito casos asociados con autismo12,13.
No se han descrito asociaciones de la trisomía 8 con presencia de quistes del plexo coroideo en el feto, y se han observado asociados a otras aneuplodías cromosómicas, como las trisomías 18 y 2114–23, por lo que presentamos un caso en el que se llegó al diagnóstico prenatal de trisomía 8 mosaico al efectuar un estudio citogenético tras amniocentesis por presencia de un quiste del plexo coroideo en la semana 18 de la gestación y un hemangioma placentario.
Paciente y métodosTercigesta de 36 años de edad, con dos cesáreas anteriores, a la que se detecta un quiste del plexo coroideo unilateral y hemangioma placentario al realizar una ecografía en la semana 18 de la gestación. Debido a la edad de la paciente y a la presencia de la alteración ecográfica, se realiza una amniocentesis para estudio citogenético.
ResultadosTras el cultivo de las células procedentes de líquido amniótico, se observa la presencia de una trisomía 8 mosaico. Este hallazgo se comprueba en un segundo frasco de cultivo, así como en sangre fetal obtenida por funiculocentesis. La comprobación de la trisomía, así como el porcentaje de células trisómicas, se estudió con la técnica de FISH (hibridación in situ con fluorescencia) utilizando la sonda Vysis Cep 8 AT-rich alpha satellite, que hibrida la región centromérica 8p11.1-q11.1, y se estudian las señales fluorescentes tanto en metafases, como en núcleos interfásicos. Pudimos comprobar tres señales, si bien el porcentaje varió desde un 80% en el primer cultivo, a un 20% en el segundo y un 8% en la sangre fetal. El cariotipo de los padres fue normal..
Después de realizar cariotipo a los padres, no se aprecian alteraciones numéricas.
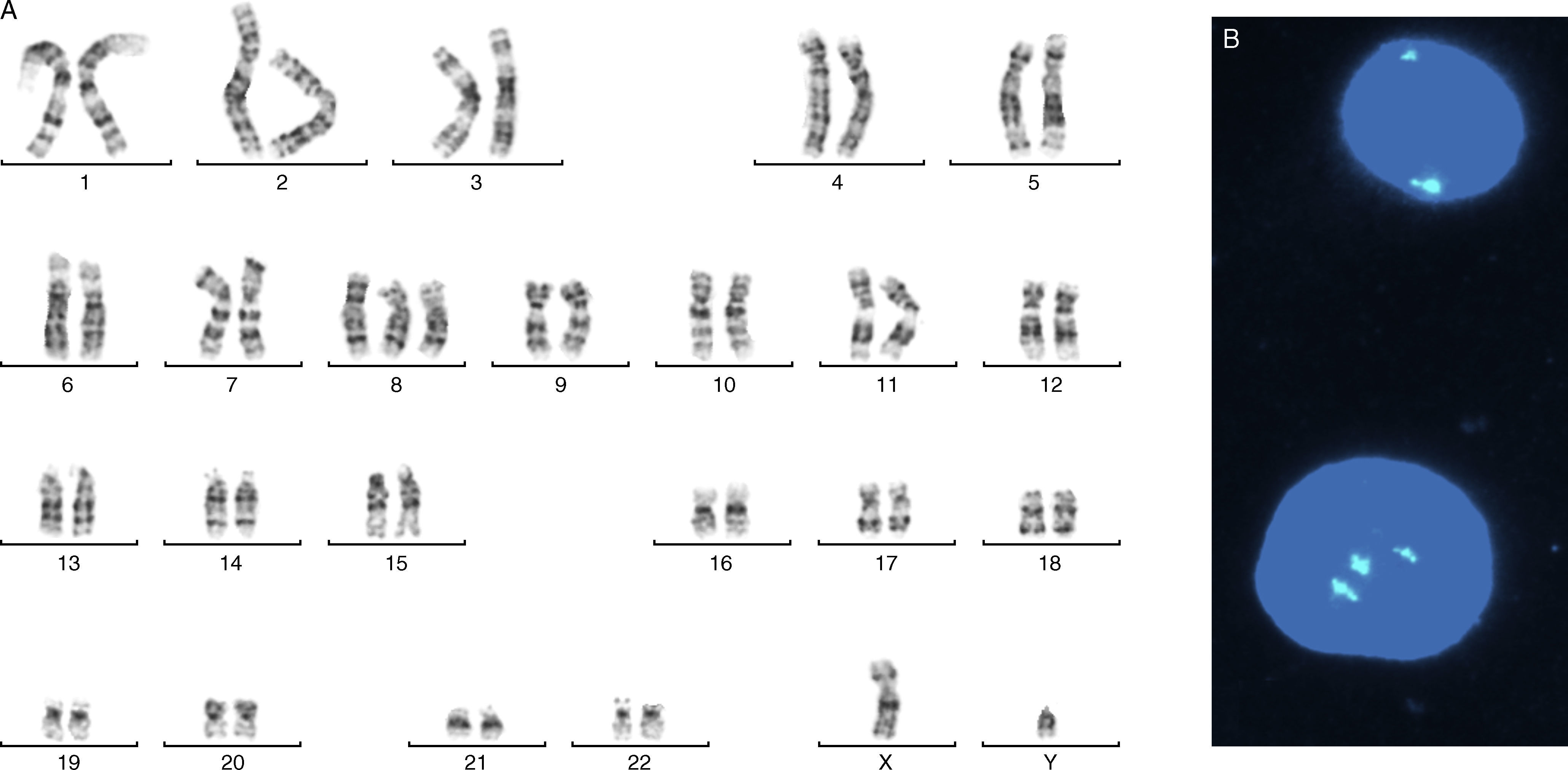
Por lo tanto, el diagnóstico queda establecido de la manera siguiente: 46, XY / 47, XY + 8. (fig. 1)
 con presencia de un núcleo con dos y otro con tres señales de hibridación con la sonda Vysis Cep 8 AT-rich alpha satellite que hibrida la región centromérica 8p11.1-q11.1.")
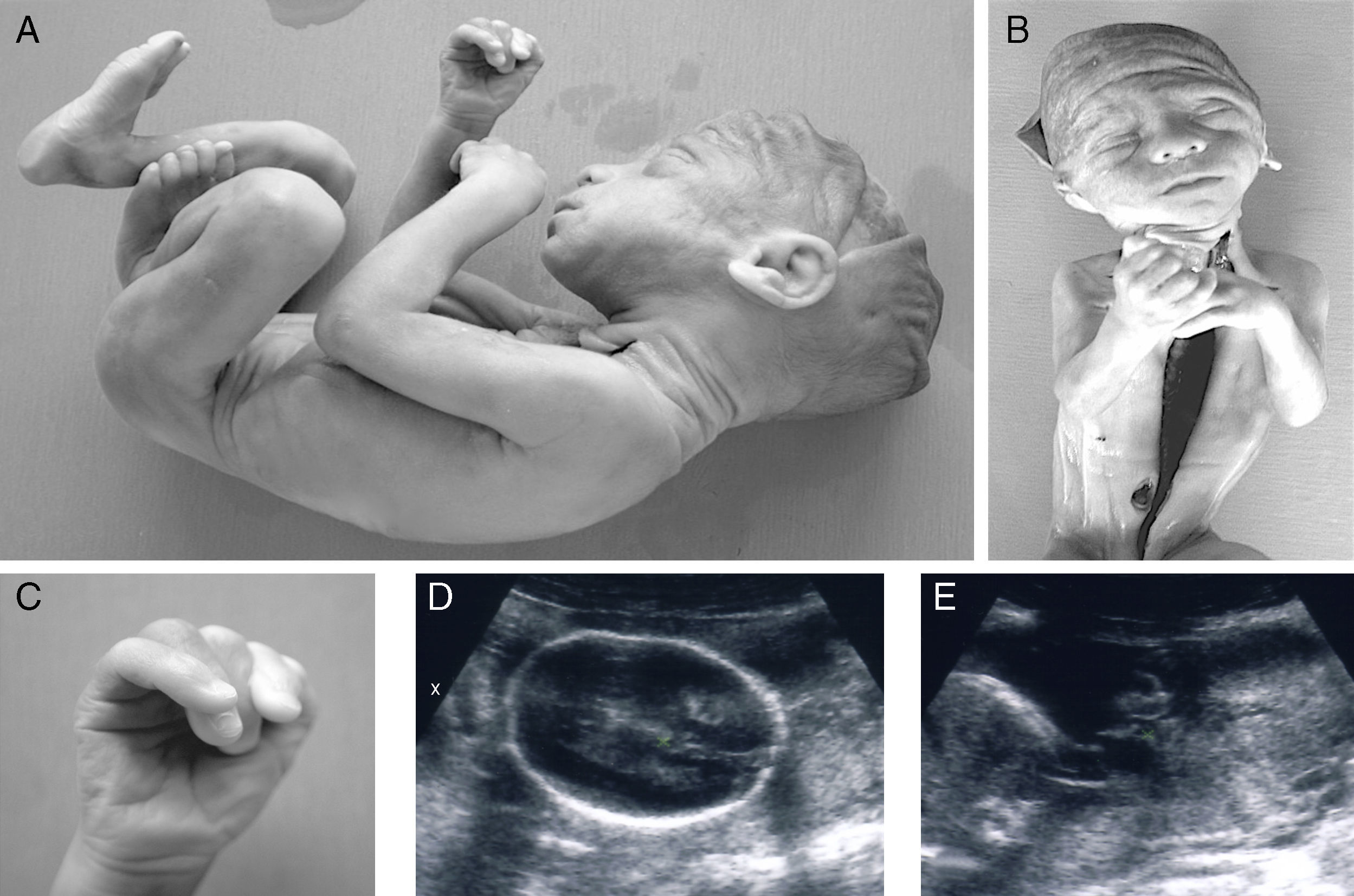
Los padres optan por la interrupción del embarazo amparándose en el tercer supuesto legal. Se realiza el estudio necrópsico, tratándose de un feto varón con una somatometría que corresponde a la 21-22 semana de gestación. Se encuentran las malformaciones externas siguientes (fig. 2 A, B, C): tórax largo, cuello corto, orejas grandes y largas de implantación baja, hombros estrechos y colgantes, palmas de las manos con surcos profundos, campodactilia (dedos de la mano derecha flexionados) y clinodactilia (1.er, 2.° y 5.° dedos de la mano derecha montados) y palas ilíacas estrechas. Asimismo, se observan signos morfológicos de anoxia intraútero —congestión visceral generalizada— y signos morfológicos de inmadurez —inmadurez pulmonar grado I, renal grado II y presencia de hematopoyesis extramedular.
Discusión
La trisomía 8, descrita por primera vez por De Grouchy et al2 en 1971, se caracteriza por una dismorfia facial muy indicativa, así como por anomalías osteoarticulares, algunas bastante graves. En los casos en los que hay un mosaicismo, la línea celular anormal tiende a desaparecer de los linfocitos con la edad, y así, en paciente adultos las aneuplodías, pueden pasar desapercibidas, a no ser que se efectúen cultivos de fibroblastos5.
Por otra parte, también hay dificultades para su diagnóstico toda vez que no existe un cuadro clínico definido, y aunque algunos síntomas son bastante constantes (tabla 1), no resultan todavía concluyentes para poder establecer un diagnóstico24–33.
Comparación de las manifestaciones clínicas en pacientes con trisomía 8 completa y trisomía 8p
| Imágenes clínicas | 47+8GROUCHI(2) | 8p12LAZJUK(30) | 8p11MATTEI(31) | 8p12JONES(29) | 8p12GEMME(28) | 8p12FRYNS(26) | 8p12RETHORE(33) | 8p12FRYNS(27) | 8p11MEMO(32) | 8p11PEZZOLO(25) | Nuestro caso |
| Dolicocefalia | + | + | + | ||||||||
| Macrosomía | + | + | + | + | + | ||||||
| Puente nasal ancho | + | + | + | + | + | + | + | + | |||
| Micrognatia | + | + | + | + | + | ||||||
| Hipertelorismo | + | + | + | + | + | + | |||||
| Orejas implantación baja | + | + | + | + | + | + | |||||
| Pterigium y/o cuello corto y ancho | + | + | + | + | + | + | + | ||||
| Clinodactilia | + | + | + | + | + | + | |||||
| Campodactilia | + | + | + | + | |||||||
| Surcos palmares y plantares profundos | + | + | |||||||||
| Labio inferior grueso y evertido | + | + | |||||||||
| Hombros estrechos y tronco largo | + | + | |||||||||
| Pie equinovaro | + | + | + | ||||||||
| Anomalías en los huesos del esplacnocráneo | + | + | + | ||||||||
| Malformación cardíaca | + | + | + | + | + | + | + | ||||
| Anomalías aórticas | + | + | + | + | |||||||
| Anomalías pulmonares | + | + | + | ||||||||
| Malformaciones renales y ureterales | + | ||||||||||
| Agenesia cuerpo calloso | + | + |
En cuanto a su etiología, Karadima et al34 realizan un estudio molecular de 26 casos, en el que se utilizan 19 marcadores de microsatélites mapeados a lo largo del cromosoma 8, y los resultados de los estudios de no-disyunción muestran que 20 casos fueron probablemente debidos a duplicación mitótica poscigótica. Solo 2 casos se debieron a una no-disyunción meiótica materna, y en 4 casos no fue posible detectar el cromosoma extra debido al bajo nivel de mosaicismo. Estos resultados están en contra de las trisomías autonómicas más comunes, en las que casi siempre se deben a errores de la meiosis materna.
La desigual distribución de las células trisómicas en los diferentes tejidos35,36 y la variabilidad de los fenotipos hacen difícil realizar un consejo genético cuando se diagnostica prenatalmente. Cuando se encuentra una trisomía 8 mosaico en amniocitos, se debe hacer una funiculocentesis con el fin de confirmar el diagnóstico. Sin embargo, la sangre de cordón fetal y los amniocitos muestran un número diferente de células con alteraciones cromosómicas37 y el hallazgo de células normales en uno de los cultivos no puede eliminar el diagnóstico de trisomía 8 mosaico, tal como ocurrió en nuestro caso en el que hubo amplias variaciones en los porcentajes de células trisómicas. Por otra parte, las imágenes clínicas menores no se detectan fácilmente mediante ecografía prenatal38.
En 1998 Morcos et al23 realizan un estudio en 7.617 pacientes y en 210 casos (2,8%) encontraron quistes de plexos coroideos. En 181 casos (86%) únicamente aparecía esta anomalía ecográfica, mientras que en los 29 restantes (14%) se asociaba con otros hallazgos ecográficos. En 2 pacientes mayores de 35 años se observaron aneuplodías: una de ellas, en las que había únicamente el quiste de plexo coroideo, tenía una trisomía 21 y otra con otros hallazgos adicionales, una trisomía 18. Otros autores han señalado la asociación entre trisomía 18 y quistes de los plexos coroideos16–18,20, si bien el riesgo de aneuploidía, en el caso de haber únicamente un quiste de plexo coroideo, no fue estadísticamente significativo, a diferencia de las aneuplodías asociadas a otras alteraciones ecográficas21. Debido a estos datos, el riesgo para un feto con quistes aislados de los plexos coroideos debe interpretarse con cautela, si bien no debe de excluirse un estudio citogenético.
En 1994 Kupferminc et al22, en un estudio retrospectivo en 9.100 mujeres embarazadas de 35 años, encuentran en 102 fetos (1,1%) quistes de los plexos coroideos. En 4 de ellos estaban asociadas otras anomalías, y en 3 de ellos, anomalías cromosómicas: 2 trisomías 18 y 1 translocación desequilibrada t(3;13). En los 98 fetos restantes, los quistes de los plexos coroideos eran hallazgos aislados y en 65 de realizó amniocentesis con presencia de 4 casos con anomalías: 2 trisomías 21, 1 trisomía 18 y 1 translocación desequilibrada t(14;21). A la vista de estos datos, el riesgo de anomalías cromosómicas en casos de quistes de plexos coroideos aislados fue de 1:25, riesgo que excede el de 1:200 de pérdida fetal tras amniocentesis y el 1:126 y 1:260 para aneuplodias y síndrome de Down, respectivamente, en pacientes de 35 años.
Estos hallazgos indican que se debe ofrecer la amniocentesis cuando se observen ecográficamente quistes de los plexos coroideos como lesión aislada y única, si bien también hay que valorar el hecho de que en ocasiones los quistes de los plexos coroideos desaparecen de modo espontáneo a las 28 semanas de la gestación, razón por la cual algunos autores14,18,21 sugieren que para la indicación de estudio citogenético el diámetro de los quistes de los plexos coroideos, bien sean unilaterales o bilaterales, sea mayor de 1cm.
El hallazgo ecográfico de un hemangioma placentario en nuestra paciente no se relaciona con la aparición de cromosomopatías. Hay estudios en los que se asocia la existencia de marcadores ecográficos fetales, con el diámetro y el número de capilares en las vellosidades coriales, y se observa que en la trisomía 18 el diámetro de las vellosidades es más pequeño y el número de capilares se encuentra reducido, mientras que en la trisomía 21 las vellosidades muestran un aumento de células trofoblásticas, así como del número de capilares con presencia de hematíes nucleados39.
Agradecemos la colaboración de los técnicos de laboratorio Fátima Martín Reyes, Soledad Cáceres García y Manuel Jerez Herrera.