


La presencia en un paciente de un fenotipo clínico característico es la indicación usual para el estudio posnatal de posible disomía uniparental (DUP). En diagnóstico prenatal la situación es más compleja, ya que la información clínica es mucho más limitada, y los estudios de DUP se plantean únicamente en determinadas situaciones de riesgo, relacionadas con la presencia de alteraciones cromosómicas numéricas o estructurales en el conjunto feto/placenta o en los progenitores, o con la detección ecográfica de algún tipo de anomalías del desarrollo fetal o de malformaciones fetales. El objetivo del presente trabajo es presentar la experiencia de nuestro centro en diagnóstico prenatal de DUP.
MétodosDesde 1998 se han realizado en nuestro centro 165 estudios prenatales de DUP de los cromosomas 6 (n=1), 7 (n=24), 11 (n=4), 14 (n=81) y 15 (n=55) correspondientes a 154 gestaciones; en 11 de ellas se han estudiado 2 cromosomas a la vez. El estudio molecular se ha realizado mediante el análisis de segregación de marcadores microsatélites polimórficos distribuidos a lo largo del cromosoma implicado.
ResultadosSe han detectado 2 casos de DUP materna, uno del cromosoma 7 y otro del 15, ambos debidos a mosaicos confinados a placenta de la trisomía correspondiente. Se ha valorado la tasa de detección de DUP con respecto al tipo de indicación.
ConclusionesAunque la DUP es un fenómeno poco frecuente, la gravedad de sus consecuencias clínicas hace absolutamente recomendable su estudio prenatal en las situaciones de riesgo definidas por las normativas internacionales.
The usual indication for postnatal uniparental disomy (UPD) testing is a characteristic clinical phenotype present in a patient. The situation in prenatal diagnosis is more complex because the clinical information is much more limited, and the UPD tests are only considered in certain risk situations related to the presence of numerical or structural chromosomal abnormalities in the foetal/placental unit and/or in the parents, or with the ultrasound detection of foetal developmental anomalies or foetal malformations. The objective of this study is to present the experience of our centre in UPD prenatal testing.
MethodsA total of 165 UPD prenatal analyses were performed out in our centre since 1998, involving chromosomes 6 (n=1), 7 (n=24), 11 (n=4), 14 (n=81), and 15 (n=55), corresponding to 154 gestations; 2 chromosomes have been studied at the same time in 11 of them. Molecular studies were carried out by segregation analysis of polymorphic microsatellite markers distributed along the involved chromosomes.
ResultsTwo maternal UPD cases of chromosomes 7 and 15 were detected, both of them due to confined placental mosaicism of the corresponding trisomy. The detection rate was evaluated with regard to the different indications.
ConclusionsAlthough UPD is a rare phenomenon, the severity of its clinical consequences makes its prenatal study absolutely advisable in the risk situations defined in the international guidelines.
La disomía uniparental (DUP) se produce cuando los 2 homólogos de un par cromosómico provienen de un solo progenitor. Se puede clasificar en isodisomía si ambos cromosomas son idénticos y heterodisomía si corresponden a los 2 homólogos del progenitor. Históricamente, este concepto fue propuesto por Engel1 en el año 1980, como posibilidad teórica de un fenómeno que ocurriría si por azar se uniera un gameto sin un cromosoma determinado (n-1) con otro disómico para el mismo cromosoma (n+1): su unión restablecería el número normal de cromosomas (2n). Algunos años más tarde, Spence et al.2 publicaron el primer caso identificado como DUP: una chica de 16 años con talla baja, cariotipo normal, afectada de fibrosis quística; los estudios moleculares demostraron que la madre era portadora de fibrosis quística, el padre era normal, y la chica tenía 2 copias idénticas del cromosoma 7 materno portador de la mutación. Sin embargo, se habían publicado con anterioridad algunos casos con anomalías cromosómicas estructurales que presentaban también DUP3–7.
IncidenciaLa incidencia de DUP de cualquier cromosoma en recién nacidos vivos se ha estimado en 1/3.5008. En la actualidad se han publicado ya más de 2.200 casos, recogidos en una base de datos de acceso libre9 (http://www.fish.uniklinikum-jena.de/UPD.html).
AlcanceAdemás de la DUP de cromosomas enteros, también existen casos de DUP parcial o segmentaria, cuando solo es una región cromosómica la que presenta DUP. En el otro extremo se encuentran los casos con DUP de todo el conjunto de cromosomas (genome wide), presente en la mola hidatiforme completa cuando la DUP es paterna o en el teratoma de ovario cuando es materna10.
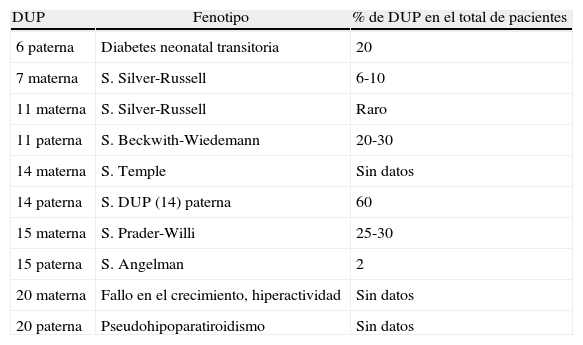
RelevanciaLa relevancia clínica de la DUP depende de varios factores: de que el cromosoma implicado presente regiones sujetas a imprinting (tabla 1), de que esté asociado a mosaicismo con una línea celular trisómica, o que provoque la homocigosidad de mutaciones recesivas (en el caso de isodisomía). El tipo de problema va a depender en parte del mecanismo de formación de la DUP.
Síndromes asociados a disomía uniparental (modificado de Yamazawa et al.28)
| DUP | Fenotipo | % de DUP en el total de pacientes |
| 6 paterna | Diabetes neonatal transitoria | 20 |
| 7 materna | S. Silver-Russell | 6-10 |
| 11 materna | S. Silver-Russell | Raro |
| 11 paterna | S. Beckwith-Wiedemann | 20-30 |
| 14 materna | S. Temple | Sin datos |
| 14 paterna | S. DUP (14) paterna | 60 |
| 15 materna | S. Prader-Willi | 25-30 |
| 15 paterna | S. Angelman | 2 |
| 20 materna | Fallo en el crecimiento, hiperactividad | Sin datos |
| 20 paterna | Pseudohipoparatiroidismo | Sin datos |
DUP: disomía uniparental; S.: síndrome.
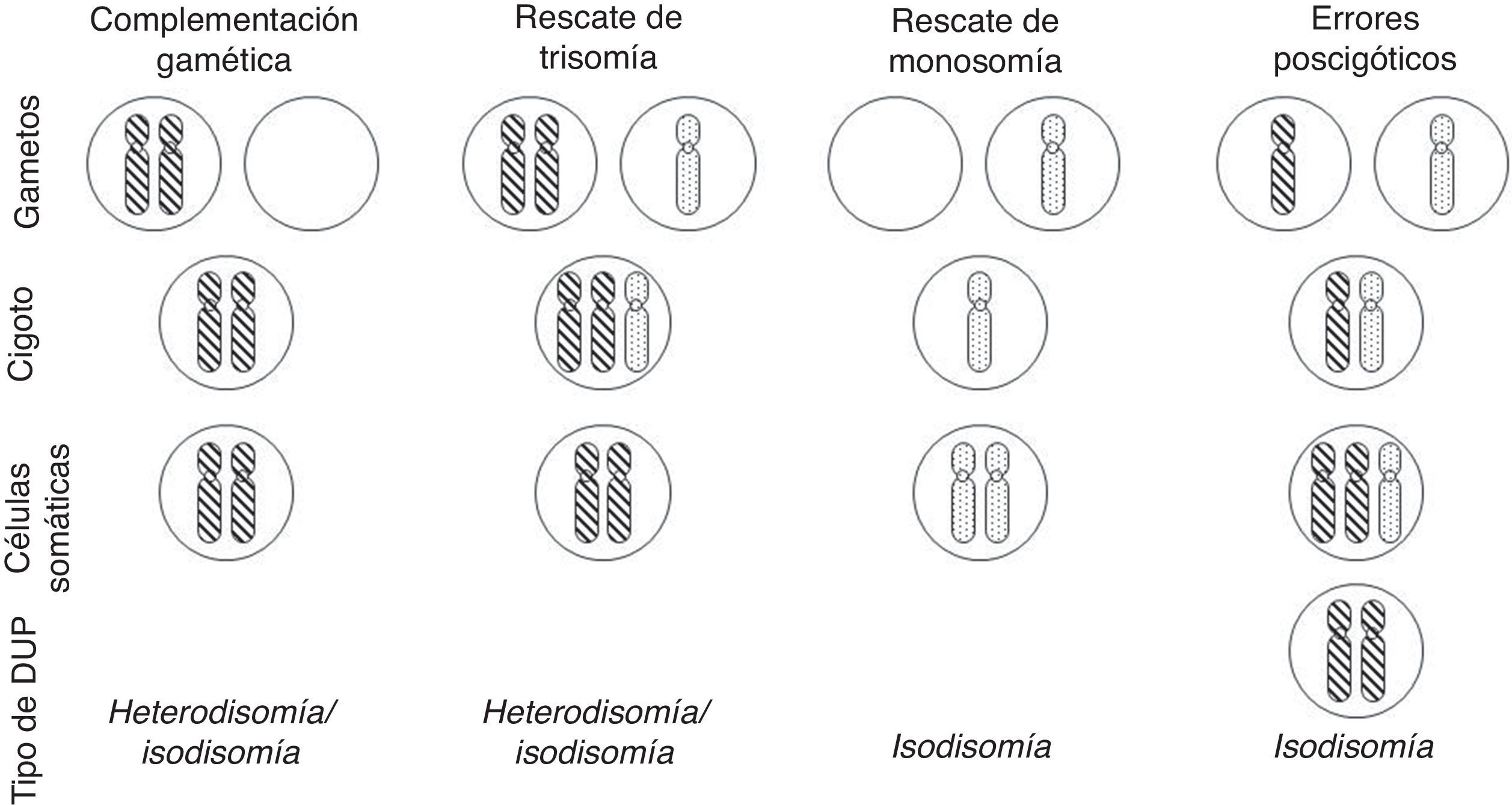
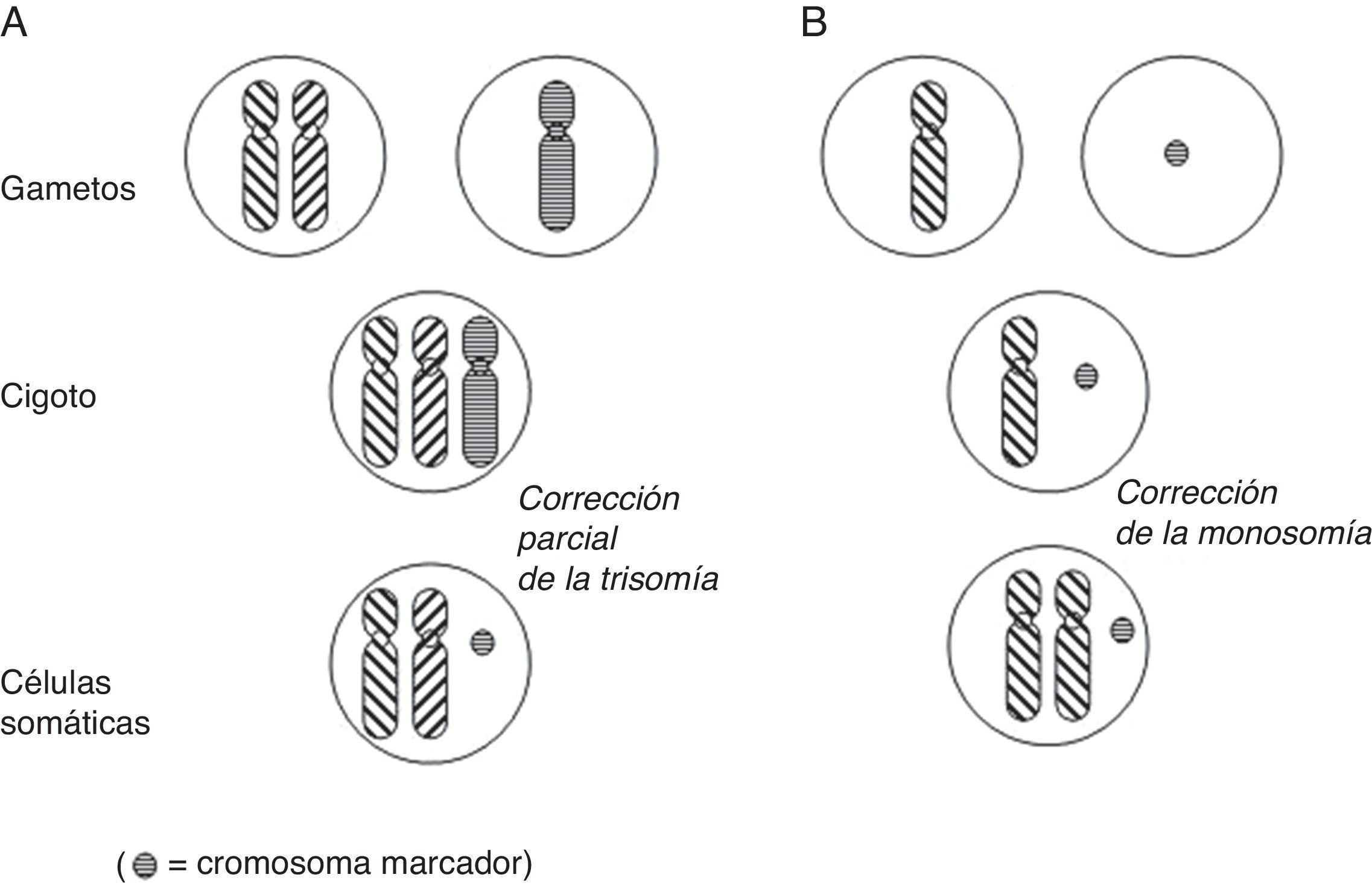
Con relación a la DUP de cromosomas enteros, existen varios mecanismos de formación: complementación gamética (el mecanismo propuesto por Engel), corrección poscigótica de una trisomía inicial, corrección de una monosomía inicial por duplicación del cromosoma implicado, o combinación de errores mitóticos poscigóticos (fig. 1).
 de cromosomas enteros (modificado de Yamazawa et al.32).")
Mecanismos de formación de disomía uniparental (DUP) de cromosomas enteros (modificado de Yamazawa et al.32).
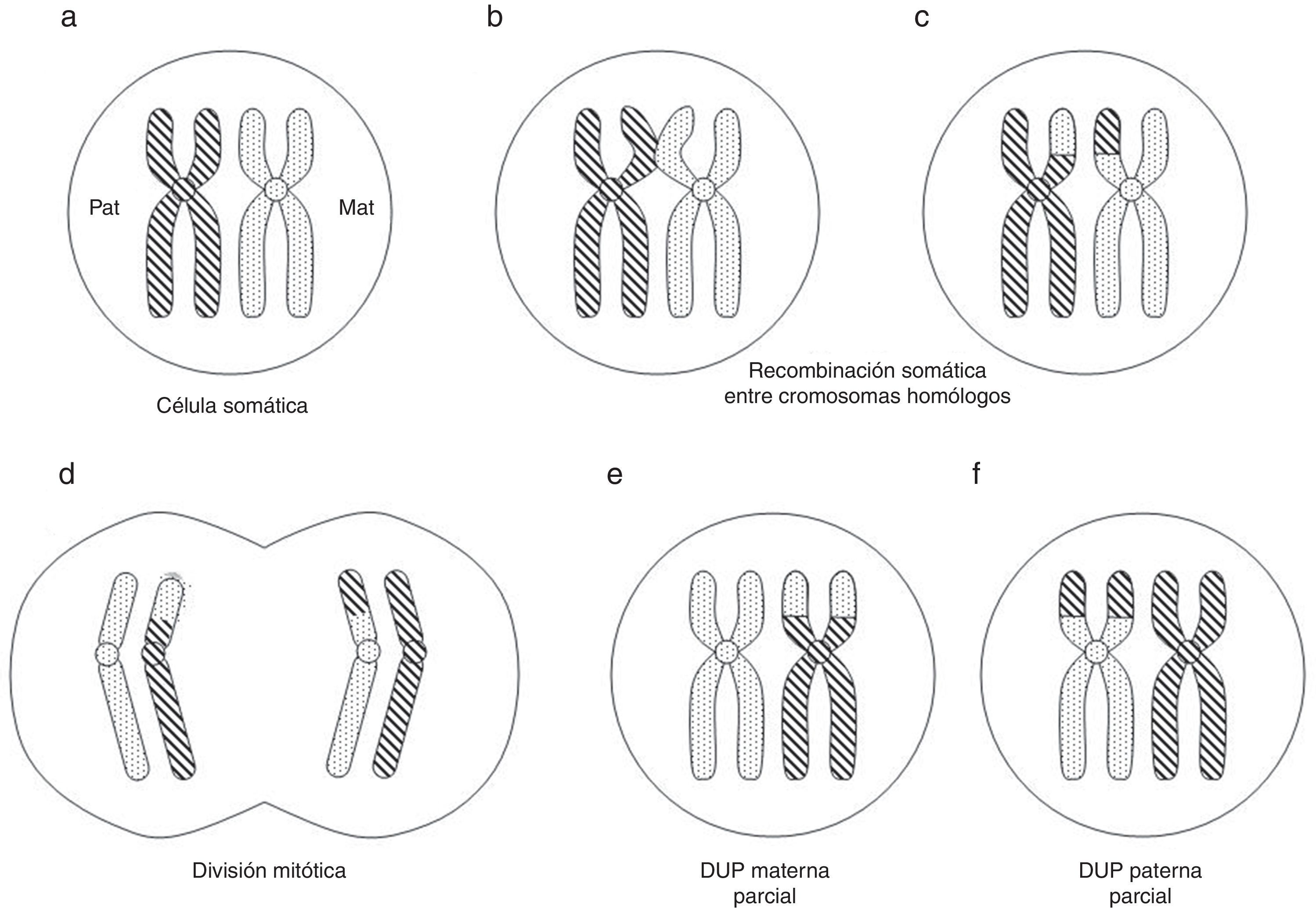
La DUP parcial o segmentaria puede producirse por recombinación somática poscigótica entre cromátidas de los cromosomas homólogos paterno y materno (fig. 2) o relacionada con anomalías cromosómicas numéricas o estructurales10.
 de disomía uniparental (DUP) parcial o segmentaria (adaptado de Gardner et al.10).")
Formación (poscigótica) de disomía uniparental (DUP) parcial o segmentaria (adaptado de Gardner et al.10).
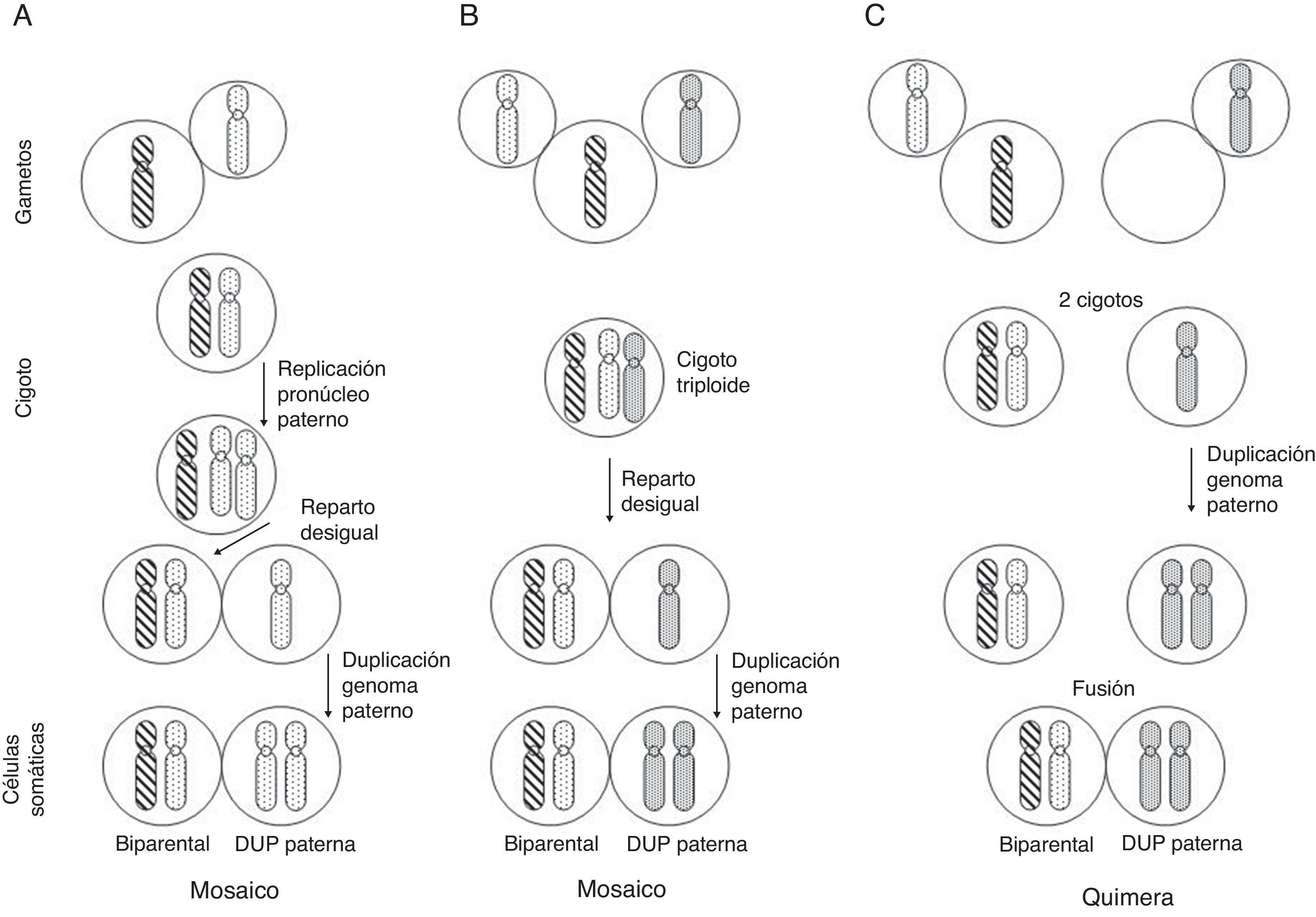
En cuanto a la DUP de todo el conjunto de cromosomas, se conocen algunos casos en los que la línea celular con DUP (paterna en prácticamente todos los casos publicados) convive con una línea celular biparental normal (mosaico/quimera androgenético/biparental), y los posibles mecanismos de formación implican o bien una fecundación normal con replicación del pronúcleo masculino y reparto desigual entre células hijas con duplicación del genoma masculino en una de las líneas celulares, o una fecundación con 2 espermatozoides seguida de un reparto desigual como en el caso anterior, o finalmente, una quimera formada por la fusión de un cigoto normal y otro resultante de la fecundación de un óvulo vacío por otro espermatozoide con duplicación del genoma masculino11 (fig. 3).
 o paterno (punteado) (adaptado de Morales et al.22).")
Posibles mecanismos de formación de mosaicismo con una línea celular biparental y otra con DUP del genoma paterno. Cada cromosoma de la figura equivale al genoma haploide materno (rayado) o paterno (punteado) (adaptado de Morales et al.22).
En los casos de DUP asociados a anomalías cromosómicas estructurales, las técnicas citogenéticas convencionales pueden contribuir a su detección, pero en general son las técnicas moleculares las que dan el diagnóstico: análisis de microsatélites, tests de metilación y microarrays de SNP. El análisis de los progenitores es necesario para la interpretación adecuada de los hallazgos.
Indicaciones para el estudio de disomía uniparentalLa presencia en un paciente de un fenotipo clínico característico (tabla 1) es la indicación usual para el estudio posnatal de posible DUP. En diagnóstico prenatal la situación es más compleja, ya que la información clínica es mucho más limitada y los estudios de DUP se plantean únicamente en determinadas situaciones de riesgo, relacionadas con la presencia de alteraciones cromosómicas numéricas o estructurales en el conjunto feto/placenta o en los progenitores, o con la detección ecográfica de algún tipo de anomalías del desarrollo fetal o de malformaciones fetales11.
Situaciones de riesgo de disomía uniparental en diagnóstico prenatalAnomalías cromosómicas numéricasEl mecanismo esencial para que se establezca una DUP es la no-disyunción meiótica, que está altamente asociada a la edad materna avanzada.
La existencia de mosaicos confinados a la placenta (MCP) en el 1-2% de las gestaciones de primer trimestre nos indica que existe un riesgo potencial de DUP producida por el mecanismo más frecuente: la corrección de una trisomía inicial. Este hecho se puso de manifiesto a posteriori con los casos de niños con síndrome de Prader-Willi nacidos tras haberse diagnosticado una trisomía 15 confinada a la placenta12,13. No todos los MCP son de origen meiótico. Se ha observado que, para determinados cromosomas, el origen más probable de los mosaicos es mitótico, en cuyo caso el riesgo de DUP es muy bajo, mientras que en el caso de mosaico de origen meiótico el riesgo de DUP es de 1/3.
Así pues, cuando en un estudio cromosómico en vellosidades coriales se detecta una trisomía de alguno de los cromosomas sujetos a imprinting (tabla 1) y el cariotipo en líquido amniótico o sangre fetal es normal, debe realizarse el estudio para descartar una posible DUP del cromosoma implicado. Algunos autores lo aconsejan solamente para los cromosomas 14 y 15, cuyos correspondiente síndromes presentan un fenotipo más grave11. Debe tenerse en cuenta, además, que la DUP puede coexistir con una línea trisómica residual que puede agravar el fenotipo clínico.
Anomalías cromosómicas estructuralesTranslocaciones robertsonianasEl hallazgo, en un diagnóstico prenatal, de una translocación robertsoniana en que esté implicado el cromosoma 14 o el 15 (ambos sujetos a imprinting), tanto entre homólogos como no-homólogos, constituye una situación evidente de riesgo de DUP con afectación clínica. Si la translocación es entre homólogos, aunque la mayoría se hayan originado posfecundación, el riesgo es más alto y, si se trata de un isocromosoma, la DUP está obviamente presente. Los estudios publicados de gestaciones con translocaciones robertsonianas no-homólogas con presencia de los cromosomas 14 o 15, tanto de novo como heredadas, dan una cifra del 0,6% de riesgo de DUP14.
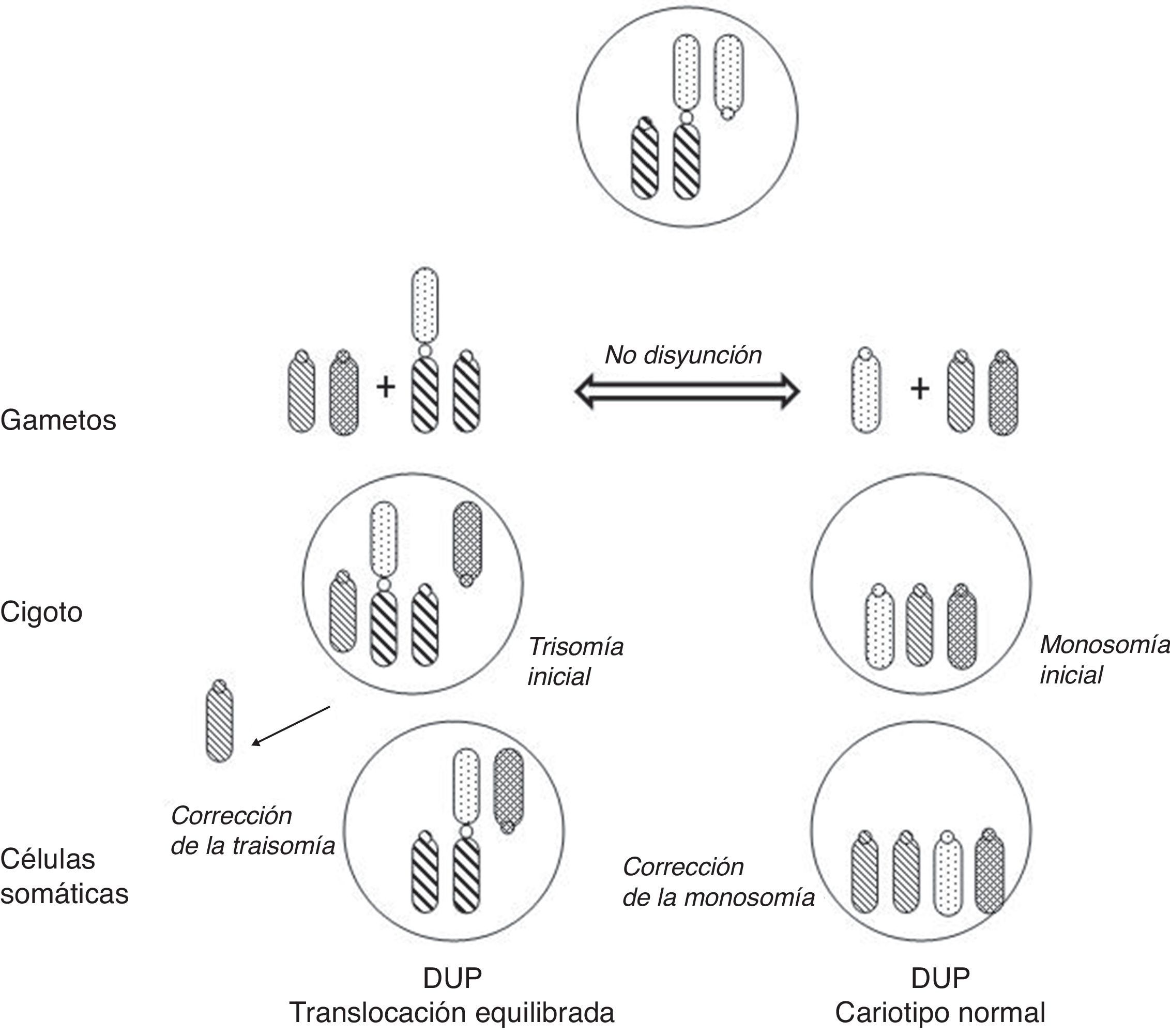
Los portadores de estas translocaciones no-homólogas tienen riesgo de DUP en su descendencia tanto si transmiten la translocación como si no puesto que, en su meiosis, una segregación desequilibrada puede dar lugar a gametos nulisómicos o trisómicos para alguno de los cromosomas implicados, y producirse la corrección de la monosomía o de la trisomía correspondiente en el cigoto (fig. 4).
.")
Mecanismos de formación de DUP en la descendencia de portadores de translocación robertsoniana equilibrada (adaptado de Kim et al.33).
Las translocaciones recíprocas que implican a alguno de los cromosomas con imprinting también son indicación de riesgo de DUP con afectación clínica, especialmente las translocaciones con probable segregación 3:1, aunque los casos descritos son muy escasos9. La mayoría de los casos publicados implican al cromosoma 1515.
Cromosomas marcadores supernumerariosLa presencia de un cromosoma marcador supernumerario en un diagnóstico prenatal puede ser indicativo de riesgo de DUP, como se ha demostrado en casos de pacientes de Prader-Willi/Angelman con un marcador inv dup(15) y disomía uniparental materna/paterna16,17. La base de datos de Liehr9 recoge actualmente 56 casos en los que la presencia de DUP está asociada a un cromosoma marcador. El estudio de una posible DUP adquiere importancia cuando el cromosoma marcador per se no tiene repercusión clínica (que esté formado únicamente por heterocromatina) y el par cromosómico correspondiente presenta imprinting. Hasta el momento, solamente se han encontrado asociados a DUP los marcadores de novo18. El mecanismo de formación más probable sería o bien la corrección parcial de una trisomía inicial en la que se perdería la mayor parte del cromosoma procedente del gameto normal, o la fecundación de un gameto normal por otro portador del marcador (debido a fragmentación cromosómica en la línea germinal), con duplicación del cromosoma íntegro (fig. 5). Curiosamente, la DUP materna es 9 veces más frecuente que la paterna en los casos asociados a marcadores18.
 la disomía puede ser iso/hetero. B) isodisomía obligada.")
Dos de los mecanismos propuestos por Liehr et al.18 para explicar la coincidencia de DUP con la presencia de un cromosoma marcador de novo. A) la disomía puede ser iso/hetero. B) isodisomía obligada.
Descartadas las malformaciones graves, que pueden justificar una interrupción de la gestación o una cirugía fetal, algunos hallazgos ecográficos pueden hacer sospechar una posible disomía uniparental en el feto. Entre ellos tenemos la restricción de crecimiento asimétrica, que podría ser indicativa de DUP materna del cromosoma 7 (síndrome de Silver-Russell), el conjunto de sobrecrecimiento fetal con macroglosia y onfalocele, que podría indicar una DUP paterna del cromosoma 11 (síndrome de Beckwith-Wiedermann), un tórax pequeño con costillas en forma de campana, que podría estar asociado a DUP paterna del cromosoma 14, o una placenta con displasia mesenquimática, que podría ser indicativa de DUP paterna de todo el conjunto cromosómico.
Recomendaciones (guidelines) internacionalesEn los últimos años varias sociedades médico-científicas de distintos países han publicado recomendaciones sobre cuándo está indicado un estudio prenatal de DUP. A continuación se describen algunas de las más recientes.
La Association for Clinical Cytogenetics (ACC, Gran Bretaña) propone considerar el estudio de DUP en: a) las gestaciones con MCP de los cromosomas 7, 11, 14 y 15; b) con cromosomas marcadores originados de los cromosomas anteriores; y c) con translocaciones robertsonianas homólogas o no-homólogas que impliquen el 14 o el 1519.
El Canadian College of Medical Geneticists (CCMG, Canadá) recomienda el estudio de DUP: a) cuando el feto presenta una translocación robertsoniana o isocromosoma, familiar o de novo, o un marcador de novo sin material eucromático, de los cromosomas 14 o 15; b) cuando el feto tiene un cariotipo normal pero uno de los progenitores es portador de una translocación robertsoniana del 14 o del 15; c) cuando se encuentra un mosaico de nivel ii o iii, en biopsia corial o en líquido amniótico, de trisomía o monosomía de los cromosomas 6, 7, 11, 14 y 15. En este último supuesto, si existe en el feto una línea celular aneuploide, el estudio de DUP puede ser clínicamente irrelevante, ya que el fenotipo del feto estará causado principalmente por la línea celular anómala. Además de estas situaciones, el CCMG propone plantearse el posible estudio de DUP en fetos con translocaciones recíprocas con intervención de cromosomas con imprinting y riesgo de segregación meiótica 3:1, y en gestaciones con hallazgos ecográficos consistentes con fenotipos de DUP20.
La European Cytogenetic Association en sus recientes recomendaciones al tratar de los estudios prenatales de DUP se remite explícitamente a las guías de la ACC21.
Presentamos a continuación la experiencia de nuestro centro con respecto a los estudios de DUP en diagnóstico prenatal.
Material y métodosDesde 1998 se han realizado en nuestro centro 165 estudios prenatales de DUP de los cromosomas 6 (n=1), 7 (n=24), 11 (n=4), 14 (n=81) y 15 (n=55) correspondientes a 154 gestaciones; en 11 de ellas se han estudiado 2 cromosomas a la vez. El 80% de las gestantes procedían de nuestro centro, y en el 20% de los casos se recibieron muestras de otros centros.
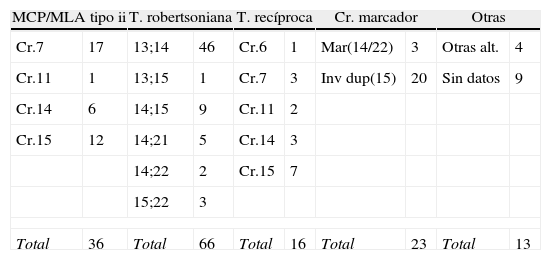
En la tabla 2 se resumen los casos estudiados según las indicaciones para el estudio de DUP.
Casos estudiados en nuestro centro distribuidos según la indicación del estudio
| MCP/MLA tipo ii | T. robertsoniana | T. recíproca | Cr. marcador | Otras | |||||
| Cr.7 | 17 | 13;14 | 46 | Cr.6 | 1 | Mar(14/22) | 3 | Otras alt. | 4 |
| Cr.11 | 1 | 13;15 | 1 | Cr.7 | 3 | Inv dup(15) | 20 | Sin datos | 9 |
| Cr.14 | 6 | 14;15 | 9 | Cr.11 | 2 | ||||
| Cr.15 | 12 | 14;21 | 5 | Cr.14 | 3 | ||||
| 14;22 | 2 | Cr.15 | 7 | ||||||
| 15;22 | 3 | ||||||||
| Total | 36 | Total | 66 | Total | 16 | Total | 23 | Total | 13 |
Para cada indicación, a la izquierda se describen los cromosomas implicados y a la derecha el n.° correspondiente de diagnósticos realizados.
MCP: mosaico confinado a placenta; MLA: mosaico en líquido amniótico.
Las muestras prenatales analizadas fueron 35 vellosidades coriales, 116 líquidos amnióticos, 2 sangres fetales y una muestra de ADN fetal sin especificar origen. En todos los casos se recogió muestra de sangre periférica de ambos progenitores. El ADN de las muestras fetales y de los padres fue obtenido mediante los protocolos habituales.
El estudio molecular se ha realizado mediante el análisis de segregación de marcadores microsatélites polimórficos distribuidos a lo largo del cromosoma que se desea estudiar de la muestra fetal y de sus progenitores. Los cromosomas analizados en el presente estudio han sido aquellos para los cuales la presencia de DUP comportaba consecuencias clínicas: 6, 7, 11, 14 y 15.
Se han utilizado primers marcados fluorescentemente (6-FAM, HEX, NET), usando los paneles ABI PRISM LINKAGE mapping de Applied Biosystems. Los productos de PCR han sido analizados en un secuenciador automático ABI3130. Para el cromosoma 15 se ha utilizado en algunos casos el kit de la casa Devyser (Devyser UPD-15). Se ha valorado la segregación alélica para varios marcadores (entre 5 y 10) del cromosoma implicado. Al menos se precisan 2 marcadores informativos para dar un resultado. En el caso de los cromosomas 7 y 15, se valoran de forma especial los marcadores incluidos en la región crítica 7q11.23 y 15q11-13.
El estudio permite determinar: si existe o no DUP, si se trata de una iso- o heterodisomía y el origen de la misma (paterno o materno).
ResultadosDel total de estudios realizados solamente se han detectado 2 casos de DUP.
El primero de ellos corresponde a una gestación con trisomía 15 completa MCP (tipo i o iii, sin cultivo largo). La gestante se sometió a una amniocentesis para comprobar la trisomía 15 hallada en el estudio semidirecto de la biopsia corial, ya que el examen ecográfico del feto era aparentemente normal, y el cariotipo del líquido amniótico fue normal. El estudio molecular mostró una DUP materna del cromosoma 15, compatible con el síndrome de Prader-Willi.
El segundo caso corresponde a una gestación con trisomía 7 en mosaico detectada en una muestra de vellosidades coriales (muestra externa, MCP sin especificar), con resultado normal en el estudio posterior en líquido amniótico. El estudio molecular mostró una DUP materna del cromosoma 7, compatible con el síndrome de Silver-Russell.
Aparte de la serie presentada, se han encontrado accidentalmente 2 casos de DUP de todo el conjunto cromosómico paterno en mosaico con una línea cromosómica biparental normal, con distintos orígenes y resultado obstétrico, que han sido previamente descritos22. En ambos casos había una discordancia entre el cariotipo fetal, normal, y la QF-PCR que mostraba un perfil de triploidía.
DiscusiónLa incidencia de DUP con afectación clínica en nuestra serie de diagnósticos prenatales ha sido baja: 2 casos positivos en 165 estudios realizados.
El riesgo de presentar DUP depende fundamentalmente de la indicación. Así, el riesgo más elevado en diagnóstico prenatal corresponde a la presencia de translocaciones robertsonianas entre homólogos e isocromosomas, establecido empíricamente por Berend et al.23 en un 66%. En nuestra serie no hay ningún caso de estos.
Siguiendo en importancia, se encuentran los MCP o los mosaicos de tipo ii o iii en líquido amniótico, siempre que el origen del mosaico sea un error meiótico y el cigoto sea inicialmente aneuploide (normalmente trisómico), en cuyo caso el riesgo de DUP es del 33,3%. Sin embargo, muchos de los MCP se deben a errores mitóticos del propio tejido placentario24, en cuyo caso el riesgo de DUP es muy bajo. Cuando en un diagnóstico prenatal nos encontramos con un mosaico de este tipo, no podemos saber su origen mitótico o meiótico si no hacemos estudios moleculares de la muestra y de los padres y, por lo tanto, el riesgo de DUP en la línea celular diploide puede estar entre < 1 y el 33,3%. Estudios realizados por diversos autores24–26 indican que existen patrones específicos para determinados cromosomas en cuanto al predominio del origen mitótico o meiótico de la trisomía correspondiente. El caso más llamativo es el de la trisomía 16, en la que predomina el origen meiótico (87%)26. Otro dato a tener en cuenta es el tipo de mosaico: los MCP tipo iii (citotrofoblasto y mesénquima anómalos) y tipo i (citotrofobasto anómalo) son los que tienen un mayor riesgo de tener un origen meiótico y asociarse a una DUP. Los CPM de tipo ii (mesénquima anómalo) son mayoritariamente de origen mitótico26,27.
En nuestra serie, de los 36 casos (tabla 2) con mosaicos hemos encontrado 2 con DUP materna, uno del cromosoma 7 y otro del 15, que representan el 5,5%. Otros estudios similares han detectado desde un 2% (1 de 51)28 a un 21% (4 de 19)27, pero este último incluía muchos casos de trisomía 16, con 3 de ellos positivos para DUP. Si nos ceñimos al cromosoma 15, cuya DUP tiene una mayor relevancia clínica, hemos encontrado un caso positivo de 12, una proporción algo menor que la publicada en el estudio colaborativo EUCROMIC29 (1 de 9) y todavía más baja que la del trabajo de Christian et al.30 (2 de 7). De todos modos el número de casos estudiados es bajo y no permite una comparación estadística. Nuestros resultados y los de Grati et al.28 coinciden en encontrar una baja tasa de DUP, que se podría explicar seguramente porque una alta proporción de los mosaicos debe tener un origen mitótico.
El riesgo de DUP asociado al cromosoma marcador más frecuente, el inv dup(15), en diagnóstico prenatal ha sido estimado en el 3,8% (2 de 26 casos)31. En nuestro estudio no hemos encontrado ningún positivo en 20 muestras.
Tampoco hemos encontrado de momento ningún caso, entre los 66 estudiados, de DUP relacionada con la presencia de translocación robertsoniana entre cromosomas no-homólogos, tanto familiar como detectada de novo en el feto. El riesgo en estos casos ha sido estimado en un 0,6%14. Aunque el riesgo sea relativamente bajo, la relevancia clínica de la DUP tanto paterna como materna de los cromosomas 14 y 15 justifica el estudio prenatal de la DUP, sobre todo cuando se ha realizado una biopsia corial o una amniocentesis por otras razones11.
ConclusiónAunque la DUP es un fenómeno poco frecuente, la gravedad de sus consecuencias clínicas hace absolutamente recomendable su estudio prenatal en las situaciones de riesgo definidas por las normativas internacionales19–21. Es sumamente importante que la gestante cuente con una información comprensible y un consejo genético adecuado.
En un futuro próximo, la aplicación de microarrays de SNP va a permitir la detección de DUP también de los cromosomas sin imprinting, con la consiguiente identificación de genes recesivos en homocigosis en los casos de iso-DUP de cualquier cromosoma.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
La parte inicial del presente estudio fue parcialmente financiada por el proyecto FIS 98/062 «Estudio de disomía uniparental y control posnatal tras el diagnóstico prenatal de mosaicos confinados a placenta».