



Se conocen numerosas reestructuraciones cromosómicas compatibles con un fenotipo normal, principalmente algunos cromosomas marcadores sin contenido genéticamente relevante y heteromorfismos cromosómicos.
Material y métodosEstudio retrospectivo de 20.098 casos prenatales.
ResultadosSe han detectado 24/17.784 casos (0,13%) de pequeños cromosomas marcadores (SMC) en líquido amniótico, 8/2.223 (0,36%) en vellosidad corial y 31/20.007 (0,15%) reestructuraciones estructurales clasificadas como heteromorfismos.
ConclusionesSe proponen guías de actuación basándose en nuestra experiencia y la bibliografía existente.
Many chromosome reorganizations compatible with a normal phenotype are known, mainly some marker chromosomes with no genetically relevant content or chromosomal heteromorphisms.
Material and methodsRetrospective study of 20,098 prenatal cases.
ResultsWe detected 24/17,784 cases (0.13%) of small marker chromosomes (SMCs) in amniotic fluid, 8/2223 (0.36%) in chorionic villus, and 31/20,007 (0.15%) structural reorganizations classified as heteromorphisms.
ConclusionsClinical practice guidelines are proposed based on our experience and the literature.
La citogenética agrupa una serie de técnicas que permiten identificar grandes mutaciones visibles al microscopio óptico como variaciones en el número, morfología o patrón de bandas de los cromosomas. Pronto se hizo evidente que las reestructuraciones cromosómicas no siempre se acompañan de efectos patogénicos sobre el fenotipo o reproductivos1. Así, se han descrito numerosos individuos sanos con pequeños cromosomas accesorios2 y variaciones de tamaño, morfología o patrón de bandas3.
Los pequeños cromosomas marcadores (small supernumerary marker chromosomes [SMC]) se definen como cromosomas que no pueden ser identificados con las técnicas de citogenética convencional y tienen un tamaño igual o inferior al cromosoma 204. Se observan en un 0,75‰ de los diagnósticos prenatales2 y se acompañan, en un contexto prenatal, de un riesgo global de anomalías fenotípicas del 26%5.
Las variaciones de tamaño, morfología o patrón de bandas que no se acompañan de efectos clínicamente relevantes se denominan «heteromorfismos»3. El uso del término «polimorfismo» se ha abandonado debido a que se refiere a características presentes en más del 1% de la población general6 y los heteromorfismos a menudo se presentan con frecuencias más bajas, o incluso están restringidos a una única familia.
La correcta caracterización de los SMC y los heteromorfismos es indispensable para determinar el riesgo real en cada caso y se basa en un conjunto de herramientas de citogenética clásica y molecular en constante evolución.
El objetivo de este trabajo es describir los principales tipos de reestructuraciones cromosómicas compatibles con un fenotipo normal y proponer unas guías básicas de actuación para su correcta caracterización en unos márgenes de tiempo y coste razonables, basándose en la bibliografía existente y el estudio retrospectivo de nuestra casuística.
Pacientes y resultadosSe ha realizado el estudio retrospectivo de 20.098 muestras prenatales (17.784 líquidos amnióticos y 2.314 biopsias coriales) recibidos en nuestro laboratorio desde el año 2007 hasta la actualidad.
En 57/17.784 muestras de líquido amniótico (0,32%) y 168/2.314 de biopsia corial (7,26%) no fue posible realizar el estudio. Mientras que las causas del fallo de crecimiento en líquido amniótico son variadas, la causa mayoritaria en biopsia corial es la ausencia de material fetal (40 biopsias), las infecciones de cultivo frecuentemente cándidas asociadas a la extracción de muestra por vía vaginal (21 biopsias) y la cantidad menor de 3mg de material fetal (30 biopsias). Excluyendo los 3 motivos de anulación, muy dependientes de las condiciones de la muestra recibida, no se pudo obtener resultado citogenético en 77/2.146 (3,3%) de las muestras. En 120 fue posible un resultado parcial mediante QF-PCR.
Se han detectado 914/17.784 alteraciones (5,13%) en líquido amniótico y 361/2.223 (16,24%) en biopsia corial.
En 24/17.784 casos (0,13%) de líquido amniótico y 8/2.223 (0,36%) de vellosidad corial la alteración era un SMC. En algunos casos no se pudo completar la caracterización (presumiblemente continuada por otros laboratorios) pero, en al menos 8 casos, se demostró compatibilidad con un fenotipo normal (3 SMC familiares y 5 derivados del cromosoma 15 sin zona crítica PW/AG y estudio de disomía uniparental negativo).
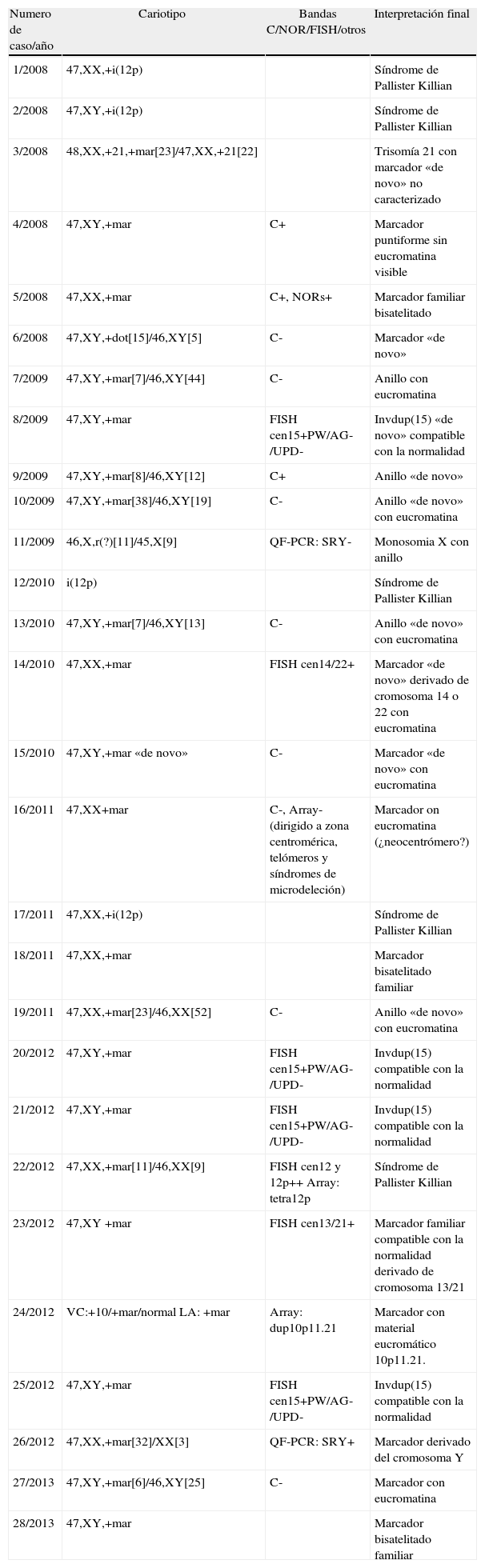
En la tabla 1 se detalla el seguimiento e interpretación de 28 marcadores. No se incluyen 2 marcadores detectados en VC no confirmados en LA y que, por tanto, se interpretaron como anomalías confinadas a placenta.
Casos de nuestra casuística en los que se ha hallado un marcador
| Numero de caso/año | Cariotipo | Bandas C/NOR/FISH/otros | Interpretación final |
| 1/2008 | 47,XX,+i(12p) | Síndrome de Pallister Killian | |
| 2/2008 | 47,XY,+i(12p) | Síndrome de Pallister Killian | |
| 3/2008 | 48,XX,+21,+mar[23]/47,XX,+21[22] | Trisomía 21 con marcador «de novo» no caracterizado | |
| 4/2008 | 47,XY,+mar | C+ | Marcador puntiforme sin eucromatina visible |
| 5/2008 | 47,XX,+mar | C+, NORs+ | Marcador familiar bisatelitado |
| 6/2008 | 47,XY,+dot[15]/46,XY[5] | C- | Marcador «de novo» |
| 7/2009 | 47,XY,+mar[7]/46,XY[44] | C- | Anillo con eucromatina |
| 8/2009 | 47,XY,+mar | FISH cen15+PW/AG-/UPD- | Invdup(15) «de novo» compatible con la normalidad |
| 9/2009 | 47,XY,+mar[8]/46,XY[12] | C+ | Anillo «de novo» |
| 10/2009 | 47,XY,+mar[38]/46,XY[19] | C- | Anillo «de novo» con eucromatina |
| 11/2009 | 46,X,r(?)[11]/45,X[9] | QF-PCR: SRY- | Monosomia X con anillo |
| 12/2010 | i(12p) | Síndrome de Pallister Killian | |
| 13/2010 | 47,XY,+mar[7]/46,XY[13] | C- | Anillo «de novo» con eucromatina |
| 14/2010 | 47,XX,+mar | FISH cen14/22+ | Marcador «de novo» derivado de cromosoma 14 o 22 con eucromatina |
| 15/2010 | 47,XY,+mar «de novo» | C- | Marcador «de novo» con eucromatina |
| 16/2011 | 47,XX+mar | C-, Array- (dirigido a zona centromérica, telómeros y síndromes de microdeleción) | Marcador on eucromatina (¿neocentrómero?) |
| 17/2011 | 47,XX,+i(12p) | Síndrome de Pallister Killian | |
| 18/2011 | 47,XX,+mar | Marcador bisatelitado familiar | |
| 19/2011 | 47,XX,+mar[23]/46,XX[52] | C- | Anillo «de novo» con eucromatina |
| 20/2012 | 47,XY,+mar | FISH cen15+PW/AG-/UPD- | Invdup(15) compatible con la normalidad |
| 21/2012 | 47,XY,+mar | FISH cen15+PW/AG-/UPD- | Invdup(15) compatible con la normalidad |
| 22/2012 | 47,XX,+mar[11]/46,XX[9] | FISH cen12 y 12p++ Array: tetra12p | Síndrome de Pallister Killian |
| 23/2012 | 47,XY +mar | FISH cen13/21+ | Marcador familiar compatible con la normalidad derivado de cromosoma 13/21 |
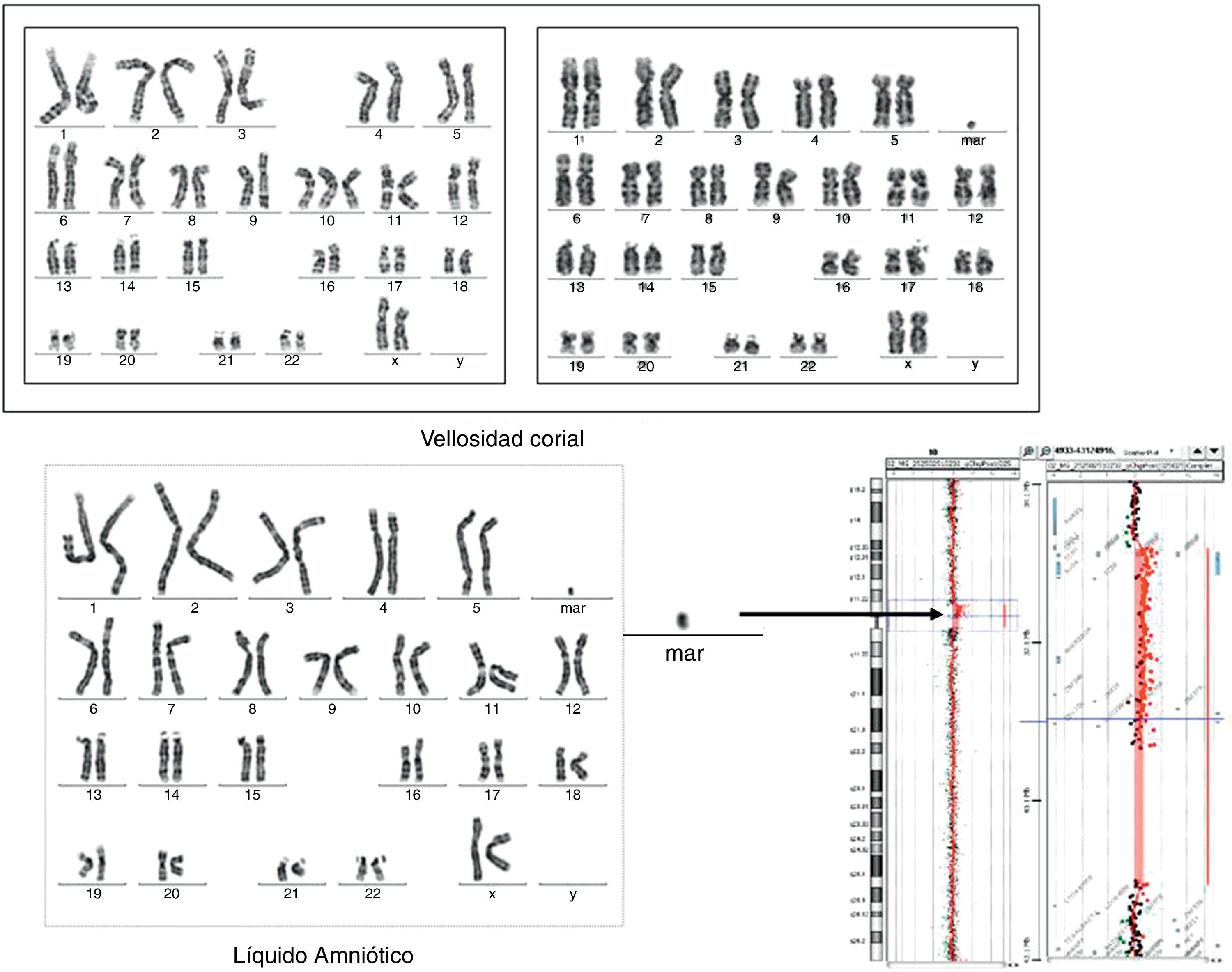
| 24/2012 | VC:+10/+mar/normal LA: +mar | Array: dup10p11.21 | Marcador con material eucromático 10p11.21. |
| 25/2012 | 47,XY,+mar | FISH cen15+PW/AG-/UPD- | Invdup(15) compatible con la normalidad |
| 26/2012 | 47,XX,+mar[32]/XX[3] | QF-PCR: SRY+ | Marcador derivado del cromosoma Y |
| 27/2013 | 47,XY,+mar[6]/46,XY[25] | C- | Marcador con eucromatina |
| 28/2013 | 47,XY,+mar | Marcador bisatelitado familiar |
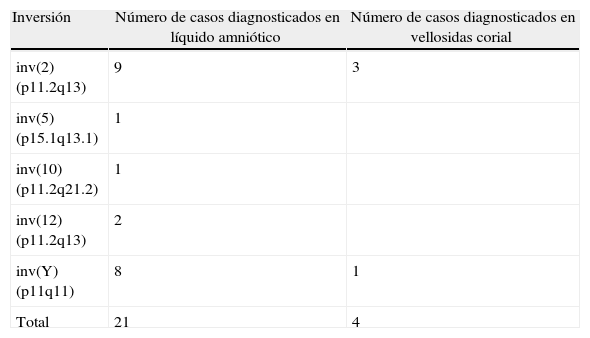
Finalmente, se han hallado 31/20.007 (0,15%) reestructuraciones estructurales clasificadas como heteromorfismos: 25 inversiones consideradas inocuas y 6 cromosomas Y satelitados (tabla 2). Debido a lo subjetivo de su detección, no se han incluido en este estudio retrospectivo las variaciones de tamaño y posición de la heterocromatina pericentromérica, los brazos pequeños ni la zona satelitar de los cromosomas acrocéntricos.
Número de casos de nuestra casuística en los que se ha hallado un heteromorfismo
| Inversión | Número de casos diagnosticados en líquido amniótico | Número de casos diagnosticados en vellosidas corial |
| inv(2)(p11.2q13) | 9 | 3 |
| inv(5)(p15.1q13.1) | 1 | |
| inv(10)(p11.2q21.2) | 1 | |
| inv(12)(p11.2q13) | 2 | |
| inv(Y)(p11q11) | 8 | 1 |
| Total | 21 | 4 |
En todos los casos analizados se demostró una herencia familiar, con la única excepción de una inversión del cromosoma 2. En ninguno de los casos tenemos constancia de la presencia de anomalías fetales atribuibles a los heteromorfismos.
Discusión y comentariosLa correcta asignación de un carácter patogénico o de variante de la normalidad a un SMC o una reestructuración cromosómica es indispensable para realizar un correcto asesoramiento genético y poder plantear técnicas de diagnóstico prenatal o preimplantacional con garantías de éxito.
La caracterización de las reorganizaciones cromosómicas se ha basado tradicionalmente en el uso de las técnicas de bandeo cromosómico G, C y NOR, con unos resultados frecuentemente ambiguos. Más recientemente, la incorporación de técnicas moleculares como FISH permitió la completa caracterización de algunos cromosomas marcadores y, desde hace muy pocos años, la generalización de los array CGH en la práctica clínica ha permitido identificar el origen y contenido génico en la mayoría de los casos7–9.
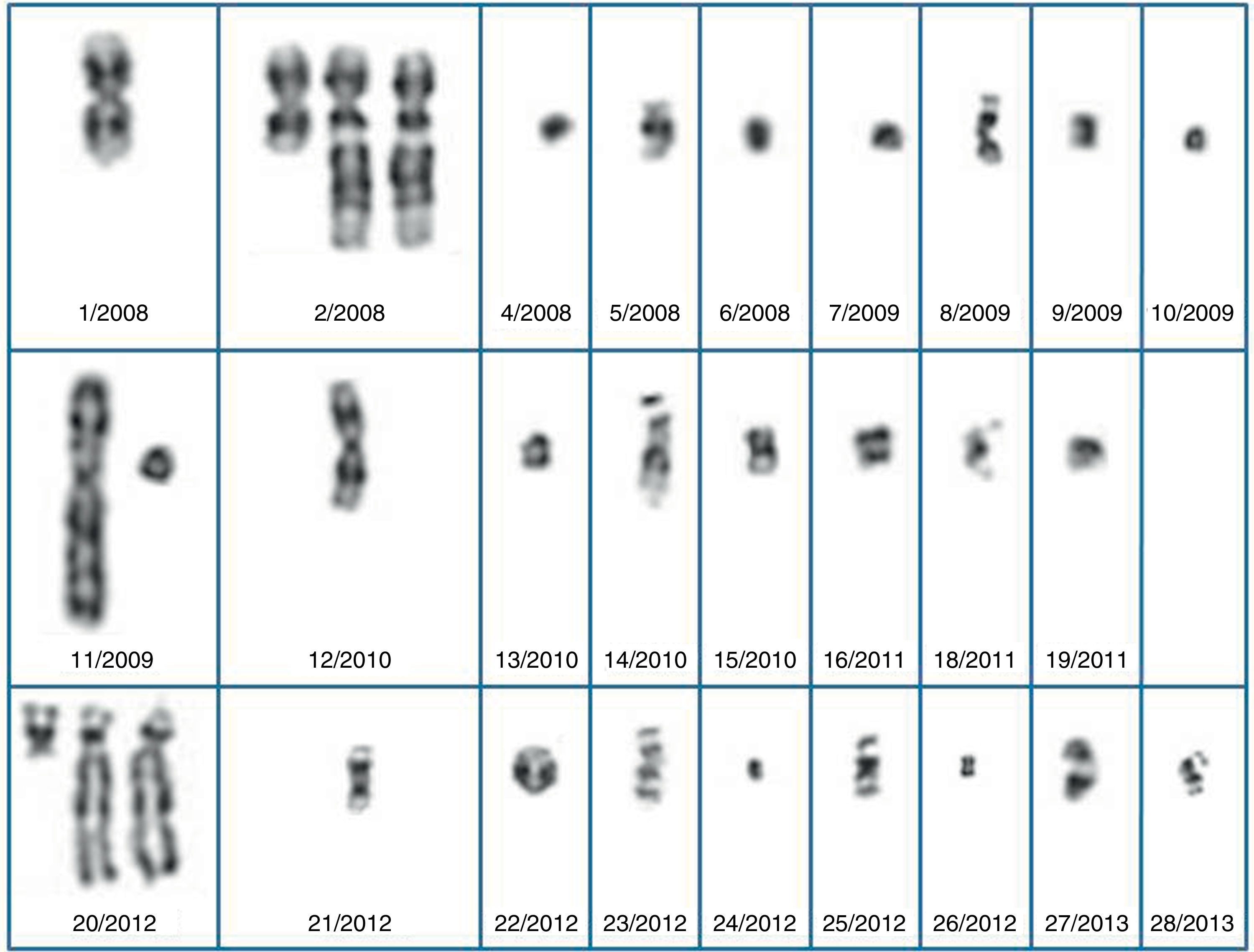
La presencia de un cromosoma marcador puede provocar anomalías fetales o ser compatible con la normalidad, en función de su procedencia y contenido génico. Por tanto, no es aconsejable el uso de cifras de riesgo globales, con frecuencia muy diferentes al riesgo real. El primer paso a seguir siempre es el estudio del cariotipo de los progenitores, incluso en los mosaicos, ya que se ha descrito la herencia a través de varias generaciones de SMC en mosaico portadores de un neocentrómero10. En caso de que exista un familiar sano portador del SMC sin evidencias de mosaico, se puede utilizar como prueba de compatibilidad con un fenotipo normal. En caso contrario, se deberá proceder a una caracterización más completa. Mediante citogenética clásica podemos diferenciar 4 tipos de marcadores: isocromosomas, marcadores bisatelitados, marcadores monosatelitados y otros marcadores menos característicos como los marcadores tipo «dot» o puntiformes (fig. 1).
Isocromosomas
Si la sospecha es firme, es recomendable intentar la confirmación con sondas FISH dirigidas, debido a su precio menor al de un array CGH y su gran sensibilidad en la detección de mosaicos. Son bien conocidos los isocromosomas 5p, 8p, 9p, 12p, 18p, Xq, Yq e Yp, que se acompañan de un fenotipo bien definido11–18.
Si la identidad del isocromosoma no es clara, la técnica de array CGH puede ser de suma utilidad, siempre que el marcador no esté en un mosaico inferior al 50%.
Marcadores bisatelitados «de novo»Si un progenitor normal es portador del marcador en forma completa (sin evidencias de mosaico) no es necesario mejorar su caracterización, ya que se ha demostrado su compatibilidad con un fenotipo normal y no son necesarios estudios de disomía uniparental, ni siquiera si procede de un cromosoma 15 o 1419.
En caso contrario, debido a que frecuentemente estos marcadores provienen del cromosoma 15, debería realizarse en primer lugar un estudio de hibridación «in situ» centromérica del cromosoma 15 y de la región 15q12.
- -
Si la prueba de FISH para el cromosoma 15 demuestra la implicación del cromosoma 15 y la presencia de señal 15q12 (zona Prader Willi/Angelman), son de esperar anomalías fetales, principalmente retraso mental.
- -
Si la prueba de FISH para el cromosoma 15 demuestra la implicación del cromosoma 15 y la ausencia de señal 15q12 se recomienda descartar una disomía uniparental:
- -
Si el resultado de la disomía uniparental es normal, el marcador es compatible con un fenotipo normal
- -
Si existe un problema de disomía uniparental, el feto presentará un síndrome de Prader Willi o Angelman.
- -
- -
Si la prueba de FISH demuestra que el marcador no deriva del cromosoma 15, se debería realizar un estudio de array CGH.
Si el array CGH no detecta anomalías, se debe asumir que se trata de un marcador sin contenido genéticamente relevante y, por tanto, compatible con un fenotipo normal. De todas maneras, debería realizarse una FISH para centrómero de cromosomas 14/22 y, en caso de ser positivo, realizar un estudio de disomía uniparental del cromosoma 14.
En nuestra experiencia, 8 de los 32 SMC detectados han sido bisatelitados (casos 5, 8, 18, 21, 22, 24, 26 y 29). Otros 4/5 SMC caracterizados con FISH resultaron proceder del cromosoma 15 y uno (caso 24) marcó con la sonda 13/21. Cabe destacar que el estudio de disomía uniparental en uno de los casos caracterizados por FISH como invdup(15) compatible con un fenotipo normal demostró la presencia de 2 cromosomas 15 procedentes del mismo progenitor compatible con un síndrome de Prader Willi.
Marcadores monosatelitados «de novo»Un marcador monosatelitado puede proceder de la parte proximal de un cromosoma acrocéntrico (caso 14) o ser un marcador complejo con material procedente de varios cromosomas20. Si un progenitor normal es portador del marcador en forma completa (sin evidencias de mosaicismo) no es necesario mejorar su caracterización, ya que se ha demostrado su compatibilidad con un fenotipo normal. En caso contrario, debería realizarse una caracterización mediante array CGH para tratar de definir el riesgo de afectación fetal.
Marcadores no satelitados «de novo»Cuando la presencia de un marcador no satelitado «de novo» se acompaña de la falta de un cromosoma sexual, la identidad más probable es la de un cromosoma X o Y. Por tanto, es recomendable realizar una hibridación «in situ» con sonda centromérica X/Y. Un marcador derivado del cromosoma Y en un paciente de fenotipo femenino se acompaña de un riesgo de gonadoblastoma y, si es muy pequeño y derivado del cromosoma X, se acompaña de un riesgo de un fenotipo Turner más severo, con retraso mental21.
En el resto de las situaciones se recomienda caracterizar el cromosoma accesorio mediante array CGH (fig. 2). En nuestra serie hemos logrado caracterizar un marcador formado por material procedente de la banda 10p11.21. Si el resultado es normal y se observa un retraso de crecimiento intrauterino es recomendable descartar una disomía uniparental del cromosoma 7. Si la técnica de FISH demuestra que el marcador procede de los cromosomas 15 o 14/22 es recomendable descartar una disomía uniparental19. Una vez caracterizado el marcador, realizar un correcto pronóstico puede no ser una tarea fácil. En estos casos, es muy importante tener en cuenta la literatura, sobre todo la web creada por Thomas Liehr (http://www.fish.uniklinikum-jena.de/sSMCl), pero existen muchos casos de marcadores con presentación en mosaico y, por tanto, su aparente compatibilidad con un fenotipo normal puede no ser real en todos los casos. En este sentido, es recomendable definir el riesgo basándonos en los casos en que el marcador ha sido hallado de forma completa.
.")
En caso de que el marcador esté en un mosaico de menos del 50% o que el marcador no esté en una región pericentromérica (probable neocentrómero) es posible que el array CGH no tenga suficiente sensibilidad. En estos casos, el aspecto citogenético con bandas G puede ser el único indicador disponible, aunque sea poco fiable. Una tinción positiva con bandas C para todo el cromosoma reduciría en riego de afectación fenotípica, mientras que la presencia de una zona blanca, indicativa de material eucromático, sería de muy mal pronóstico.
Marcadores tipo «dot» «de novo»En este grupo englobamos aquellos marcadores de tamaño similar a los satélites de los cromosomas acrocéntricos. Suelen presentar un color oscurecido por bandas G y las bandas C están al límite de poder aclarar la presencia de contenido génico. En caso de presentarse en un mosaico elevado o total, se recomienda realizar una técnica de array CGH. En otro caso, debe asumirse un riesgo de anomalías fenotípicas igual o inferior al 5%22.
HeteromorfismosDebido a lo ambiguo de los criterios de identificación, ausencia de relevancia clínica y riesgo de interpretación errónea por personas sin suficiente formación citogenética, la European Cytogeneticists Association (ECA) recomienda no incluirlos en los informes citogenéticos23,24. Este ejercicio de pragmatismo es de fácil aplicación en heteromorfismos bien descritos, como la inversión heterocromática del cromosoma 9, pero puede ser complicado de aplicar en características infrecuentes para las que pueden existir discrepancias de interpretación entre los diferentes laboratorios y puede existir cierto riesgo de confusión con variantes patogénicas morfológicamente similares. En estos casos, es recomendable explicar su probable carácter de variante de la normalidad e indicar de forma clara el protocolo a seguir (por ejemplo, tratar de confirmar la compatibilidad con un fenotipo normal mediante el estudio de los progenitores).
Variaciones en la intensidad de tinciónLa técnica de bandas Q revela con frecuencia variaciones de tinción en los cromosomas 3, 4, 13, 14, 15, 21 y 22 en sujetos normales25,26. Este tipo de variación es poco o nada visible con otras técnicas de bandas, y la sustitución de las bandas Q por las G en la rutina diagnóstica ha significado su olvido casi total.
Variaciones de tamaño en la heterocromatinaLa heterocromatina es un tipo particular de cromatina altamente repetitiva y densamente empaquetada27 en la cual son muy frecuentes las variaciones de tamaño, especialmente en los cromosomas 1, 9, 16 e Y. Con toda probabilidad, la heterocromatina pericentromérica de cualquier cromosoma puede sufrir variaciones normales de tamaño, pero han sido poco descritas en la literatura debido a su ausencia de asociación con efectos fenotípicos y a lo subjetivo de su identificación. Este tipo de variación podría ser más frecuente en los cromosomas 428, 17 (observación personal), 1829, 1930 y 2031.
En caso de duda, la técnica de bandas C es de utilidad para confirmar la implicación de la heterocromatina en la variación de tamaño y para minimizar el riesgo de confusión con una variante patogénica. Más recientemente, la técnica de array CGH permite descartar con total seguridad la presencia de duplicaciones de material eucromático.
Variaciones en los brazos pequeños de los cromosomas acrocéntricosLas variaciones de tamaño en las bandas p11, p12 y p13 de los cromosomas acrocéntricos son extremadamente frecuentes, siendo generalmente consecuencia de diferencias en el número de repeticiones de las secuencias de ADN satélite tipo i, ii, iii y iv, genes ribosómicos y secuencias beta satélite32.
Sin embargo, se han descrito incrementos en el tamaño de estas bandas como consecuencia de translocaciones crípticas y, por tanto, con efectos patogénicos33,34. La diferenciación de un incremento de tamaño patológico de una variante de la normalidad por métodos de citogenética clásica es claramente una de las limitaciones de la técnica, siendo un tema muy espinoso y de interpretación muy subjetiva. La técnica de bandas nucleolus organizing region (NOR) no es útil, ya que la translocación críptica puede ser distal a la de las estructuras NOR y, como norma general, deben preocupar los grandes aumentos de color blanco (color de la eucromatina) aunque no es infrecuente la presencia de una fina banda blanca en satélites «normales» de gran tamaño y color negro. En caso de duda, es conveniente el estudio de los progenitores, analizando con especial atención los telómeros de otros cromosomas, y valorar la utilidad de técnicas como el FISH o MLPA subteloméricas o el array CGH.
Presencia de estructuras nucleolus organizing region en cromosomas no acrocéntricosEste tipo de variación se origina como consecuencia de una translocación entre un cromosoma acrocéntrico y otro cromosoma. Las duplicaciones y deleciones de los brazos pequeños, estructuras NOR y regiones satelitares de los cromosomas acrocéntricos son compatibles con un fenotipo normal, como demuestra la existencia de portadores sanos de translocaciones robertsonianas (deleción) y SMC bisatelitados (triplicación). Por tanto, el tema se reduce a si la translocación afecta a material genéticamente relevante del cromosoma no acrocéntrico.
Una heteromorfismo muy frecuente son los cromosomas Y con satélites en el extremo de la zona heterocromática del brazo largo o Yqs. Las 2 regiones implicadas carecen de contenido génico relevante y, por tanto, es imposible que se produzca ningún efecto fenotípico. Su frecuencia es especialmente elevada en algunas poblaciones.
Si se observa la presencia de satélites en la zona distal de otros cromosomas, es conveniente realizar un estudio de array CGH si el paciente presenta anomalías fenotípicas (y por tanto se sospecha un desequilibrio cromosómico) o un estudio de FISH subtelomérico para el cromosoma implicado si el fenotipo del portador es normal y, por tanto, se sospecha que es una anomalía equilibrada. Se conocen casos en los que la translocación es tan distal que no implica cambio de material relevante y se hereda como una variante de la normalidad35,36, y casos en los que implica material relevante y, por tanto, presentan los riesgos reproductivos. La presencia de estructuras NOR intersticiales37 debe sospecharse siempre cuando se observa la presencia de un espacio vacío (gap) en todas las metafases (los sitios frágiles se presentan solo en parte de las metafases), siendo muy útil la técnica de bandas NOR como confirmación. En estos casos es recomendable la técnica de array CGH si el paciente presenta algún tipo de fenotipo, a fin de detectar pérdidas o ganancias de material, y podría ser recomendable un pintado cromosómico para descartar la presencia de material del cromosoma reorganizado en otro cromosoma. Debido a la baja resolución del pintado cromosómico, un resultado negativo rebaja, pero no excluye totalmente el riesgo de una anomalía equilibrada que determinaría un riesgo de descendencia cromosómicamente desequilibrada.
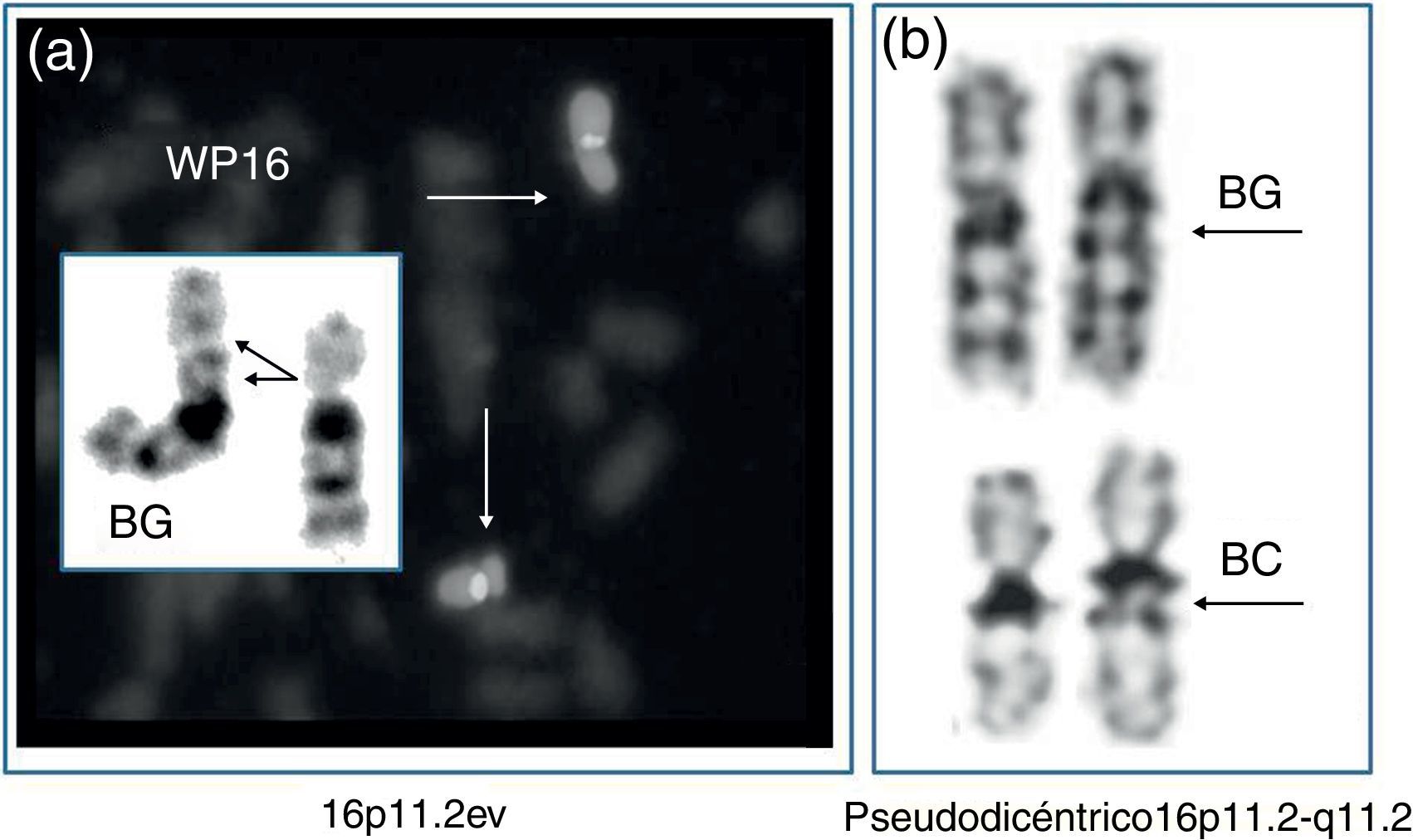
Variantes eucromáticasLas anomalías eucromáticas mas frecuentes han sido descritas por Barber38 e incluyen las variantes 8p23.1v, 9p12v, 9q12v/9qhv, 15q11.2v y 16p11.2v y la variante pseudodicéntrica de 16p11.2-q11.2 con eucromatina de 16p11.2-p11.139. En todos los casos, el estudio de array CGH puede ser definitivo para confirmar el carácter normal o patogénico de la variante. No hemos detectado ninguna de estas variantes en estudios prenatales. Con fines didácticos, la figura 3 muestra 2 ejemplos detectados de forma posnatal.
Inversiones Bandas G (BG) y pintado cromosómico (WP16) de un 16p11.2ev. b) Bandas G (BG) y C (BC) de un pseudodicéntrico 16p11.2-q11.2.")
Generalmente las inversiones pericéntricas se acompañan de un riesgo de descendencia cromosómicamente desequilibrada como consecuencia de la formación de recombinantes durante la meiosis y de anomalías fenotípicas si son «de novo», como consecuencia de disrupciones génicas o duplicaciones/deleciones crípticas40.
Sin embargo, se conocen excepciones como la inversión pericéntrica de la región heterocromática del cromosoma 9 [inv(9)(p11q13) e inv(9)(p12q13)] presente en el 3,57% de la población41 y que no se acompaña de riesgos reproductivos42. Una situación similar se observa con la inversión del cromosoma 2 [inv(2)(p11.2q13)]43.
El mecanismo molecular que origina las inversiones de los cromosomas 2 y 9 implica duplicaciones segmentales44,45. Por tanto, se pueden producir con alguna frecuencia «de novo», especialmente la inversión del cromosoma 2 (1/91)43, pero los puntos de rotura no se distribuyen al azar. Por tanto, no es de esperar un incremento de riesgo de anomalía si la inversión es «de novo» y no es previsible que el estudio de los progenitores aporte información útil. En cambio, conduce a una inevitable angustia en los casos «de novo».
No se recomienda incluir las inversiones del cromosoma 9 en los informes citogenéticos23,24, pero en el caso de la inversión 2 falta una directiva específica, por lo que se pueden producir discrepancias entre laboratorios. Por tanto, a fin de evitar confusiones, es recomendable informar la inv(2)(p11.2q13), detallando claramente que se trata de una variante de la normalidad sin efectos clínicamente relevantes.
Finalmente, no se ha demostrado la aparición recurrente en el resto de las inversiones heteromórficas inv(10)(p11.2q21.1), inv(5)(p13q13) e inv(12)(p11.2q13). En estos casos es recomendable el estudio de los progenitores a fin de permitir dar una garantía adicional de falta de efectos fenotípicos (fig. 4).
Responsabilidades éticasProtección de personas y animales.")
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.