


El carcinoma de células de Merkel (CCM) es un tumor infrecuente de la capa basal de la epidermis o de la dermis relacionado con la exposición solar y con el sistema inmune. El riesgo de padecer CCM es mayor en personas con trasplante renal que en la población general. Además, en esta situación aparece en edades más tempranas, su localización es múltiple, presenta mayor agresividad y se asocia con otros tumores de la piel y con neoplasias hematológicas. Presentamos el caso de un varón de 68 años con trasplante renal que desarrolla múltiples carcinomas epidermoides y un CCM con metástasis ganglionares regionales múltiples a los 4 años de recibir terapia inmunosupresora.
Merkel cell carcinoma (MCC) is an infrequent tumor of cells of the basal layer of the epidermis or dermis and is related to sun exposure and the immune system. The risk of developing MCC is greater in renal transplant recipients than in the general population. In these patients, this tumor develops at younger ages and in multiple locations; in addition, this entity is more aggressive, more frequently recurs after treatment, and is associated with other tumors of the skin and with hematologic neoplasms. We describe the case of a 68-year-old male renal transplant recipient who developed multiple epidermoid carcinomas and MCC with regional metastasis after 4 years of receiving immunosuppressive therapy.
El carcinoma de células de Merkel (CCM), también conocido como merkeloma o carcinoma neuroendocrino de la piel, es un tumor dérmico muy poco habitual1 que ocasionalmente puede afectar a las mucosas.
Aparece más frecuentemente en edades avanzadas y afecta en especial a personas con inmunosupresión tras trasplante de órgano sólido, en cuyo caso, su aparición suele ser más precoz2.
Su curso clínico es agresivo, con tendencia a la recidiva local, ganglionar y a distancia.
El CCM se caracteriza por la presencia de nódulos cutáneos indoloros de crecimiento rápido y de aparición más frecuente en zonas expuestas al sol.
Aunque el diagnóstico diferencial puede ser dificultoso, las peculiaridades histológicas y el perfil inmunohistoquímico del CCM son característicos3,4.
El tratamiento del CCM es similar con independencia de que afecte a pacientes con o sin inmunosupresión. Consiste en una exéresis amplia del tumor, con linfadenectomía en casos de afectación ganglionar clínica5,6. Existe disparidad de criterios respecto a la linfadenectomía en ausencia de afectación clínica. La radioterapia adyuvante puede ser útil especialmente en casos de resecciones con bordes afectos o ante imposibilidad de cirugía7,8. La quimioterapia puede utilizarse en supuestos de enfermedad diseminada.
Caso clínicoVarón de 68 años sometido a trasplante renal 4 años antes y en tratamiento inmunosupresor desde entonces con ciclosporina, azatioprina y prednisona. Había sido intervenido en diversas ocasiones por otros tantos carcinomas espinocelulares de piel.
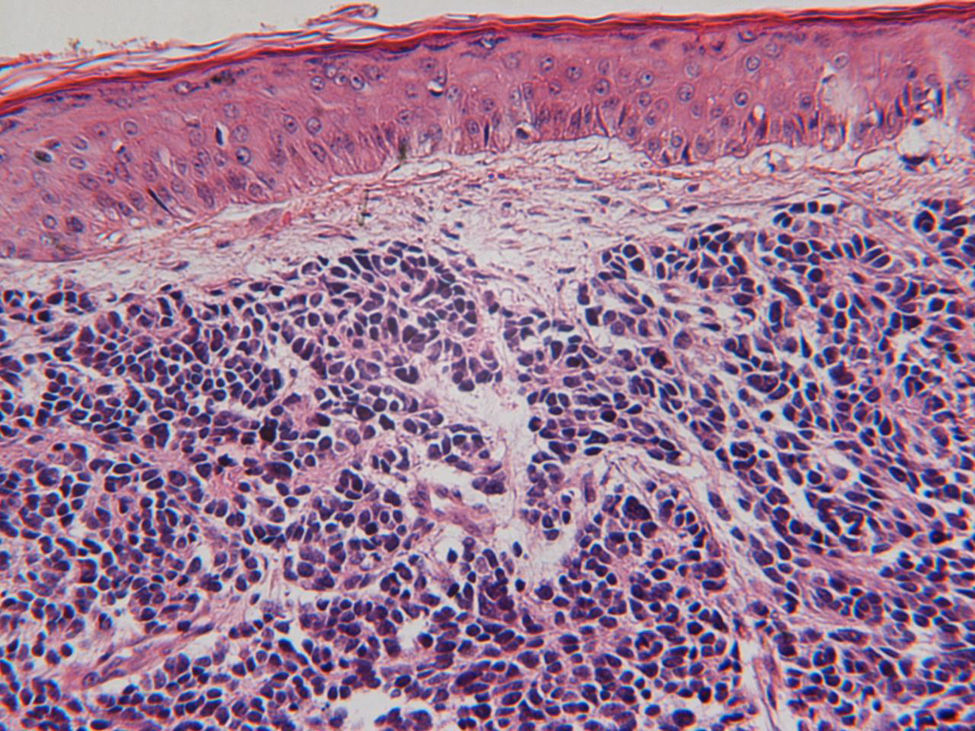
Consulta por aparición, en los tres meses previos, de una lesión eritemato-violácea de 4cm y consistencia firme en región infraclavicular izquierda. En la exploración física destacaba la presencia de una adenopatía de 3cm en axila izquierda, móvil y no dolorosa, así como varias zonas eritematosas con erosión superficial, alguna de ellas verrugosa, en región frontal izquierda, antehélix izquierdo, mejilla derecha y región supraclavicular izquierda. El examen anatomopatológico de las lesiones faciales y supraclaviculares izquierdas fue diagnóstico de carcinomas espinocelulares con bordes de resección libres. El examen microscópico de la lesión infraclavicular, cuyos bordes de resección estaban libres, mostró células con patrón de crecimiento sólido y difuso en sábana, con infiltración de la dermis papilar y reticular, ulceración de la epidermis y penetración en el tejido celular subcutáneo. Sus bordes estaban mal delimitados y existía invasión de numerosos vasos linfáticos en la periferia del tumor. Las células eran pequeñas, con citoplasma escaso y mal delimitado, elevada relación núcleo/citoplasma, núcleos ovales, pleomorfos y atípicos, sin nucleolo aparente, con cromatina dispersa en pequeños grumos (fig. 1) y numerosas mitosis que ocasionalmente formaban pequeñas seudorosetas.

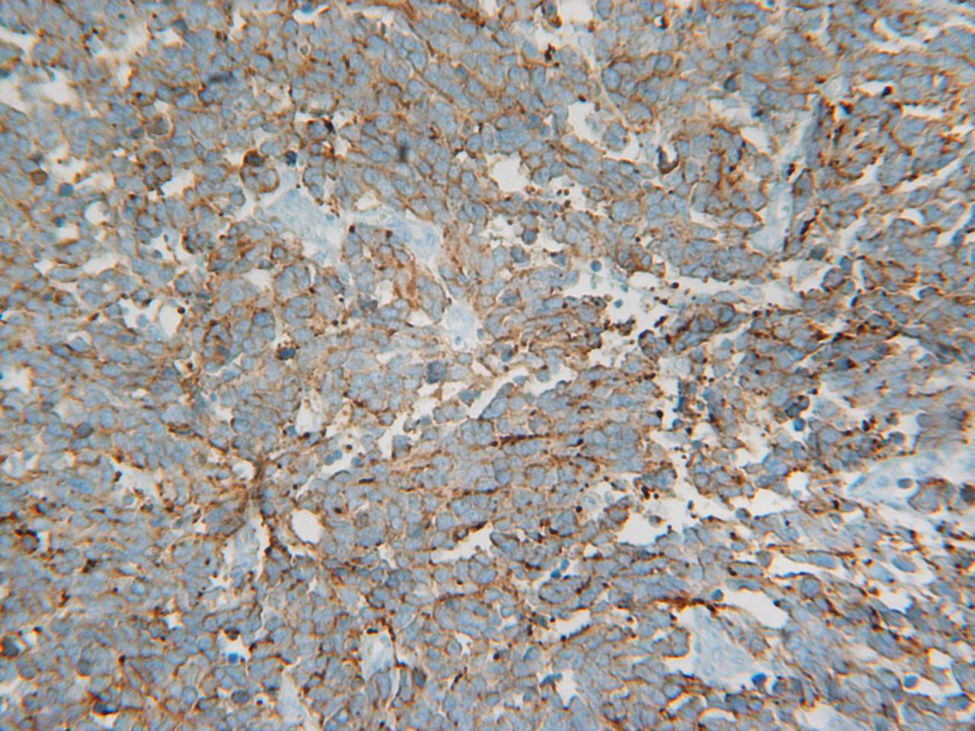
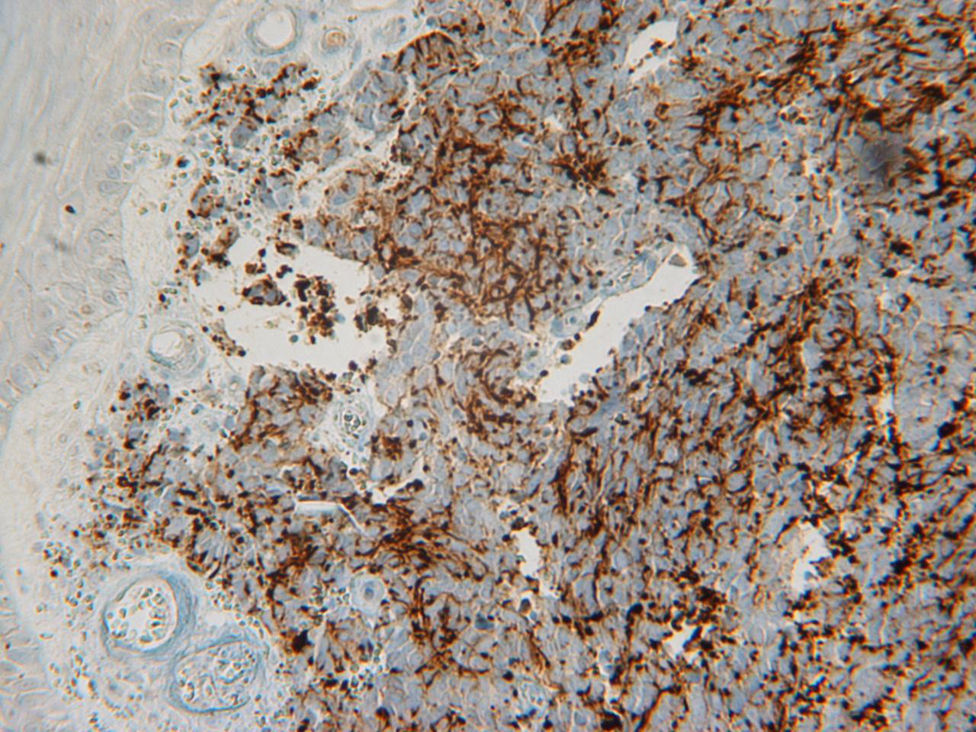
El estudio inmunohistoquímico mostró positividad de las células frente a queratina AE1/AE2, queratina CAM 5.2 y cromogranina A, y negatividad frente a CD 45. De igual modo, mostraron positividad para citoqueratina 20 (CK 20) (fig. 2) y sinaptofisina citoplasmática (fig. 3) con negatividad para TTF-1.


Tras vaciamiento axilar se confirmó el diagnóstico de carcinoma neuroendocrino cutáneo de células pequeñas o carcinoma de células de Merkel con metástasis ganglionares regionales múltiples (estadio ii), que se acompañaba de los carcinomas epidermoides con bordes de resección libres ya descritos. Se descartó, mediante TC toraco-abdomino-pélvico, la existencia de metástasis a distancia.
Se inició radioterapia adyuvante y se retiró progresivamente la inmunosupresión. Al tiempo se inició tratamiento con cisplatino y etopósido, el último ciclo de los cuales no pudo completarse por trombopenia grave. Tras varios meses de estabilidad clínica presentó una bronconeumonía con insuficiencia respiratoria aguda y grave deterioro de la función renal, que condicionó su fallecimiento.
DiscusiónLas células de Merkel derivan de la cresta neural, actúan como mecanorreceptores cutáneos e intervienen en el tacto y en el movimiento direccional del pelo.
El carcinoma de células de Merkel es un tumor infrecuente, muy agresivo, de mal pronóstico y con gran tendencia a la recidiva local. Supone menos del 1% de los tumores cutáneos malignos, con una incidencia total de 0,44 casos por 100.000 habitantes/año, mayor en varones de raza blanca, edades avanzadas y estados de inmunosupresión. En pacientes con trasplante renal, el riesgo de padecer CCM es de 0,13/1.000 pacientes trasplantados/año y la edad media de aparición es unos 10 años inferior a la de los pacientes no inmunocomprometidos1. Además, el momento de la aparición del tumor parece ser dependiente de la edad: 8 años postrasplante en pacientes de 40 años y 3 años postrasplante en mayores de 608.
La relación del CCM con el sistema inmune viene contrastada por la existencia de remisiones espontáneas mediadas por inmunidad, por su mayor prevalencia y su aparición más precoz en pacientes inmunodeprimidos crónicos, por su concordancia con el grado de inmunosupresión, traducida por recuentos de linfocitos CD 4, y por su relación con la duración, dosis y tipo de terapia inmunosupresora1,3,9.
El CCM se manifiesta clínicamente como nódulos cutáneos de 0,5 a 5cm, de crecimiento rápido, indoloros, firmes, violáceos, rara vez ulcerados, de aparición más frecuente en áreas expuestas al sol.
Su diagnóstico diferencial es amplio, por lo que son necesarias técnicas complementarias como la microscopía electrónica y la inmunohistoquímica. Es característica del CCM la tríada formada por un núcleo vesicular con pequeño nucléolo, abundantes mitosis y apoptosis. Ya se ha dicho que las técnicas inmunohistoquímicas facilitan el diagnóstico diferencial, especialmente con las metástasis cutáneas del carcinoma de células pequeñas de pulmón. El CCM expresa citoqueratinas, especialmente la CK 20, y es negativo para citoqueratina 7 (CK 7), a diferencia de lo que ocurre en los carcinomas de células pequeñas de pulmón. La expresión de neurofilamentos a modo de bola paranuclear, que ocurre en el CCM, es a menudo negativa en los carcinomas microcíticos pulmonares. Ambos tumores son positivos para enolasa neuronal específica y, ocasionalmente, para CD 99 de expresión citoplasmática. Otras entidades a considerar son el linfoma (con negatividad para citoqueratinas y positividad para antígeno leucocitario común y marcadores de linfocitos B o T correspondientes), el melanoma de células pequeñas (con negatividad para citoqueratinas y positividad para proteína S-100) y el tumor neuroectodérmico primitivo (con negatividad para citoqueratinas y positividad para enolasa neuronal específica, y CD 99 con tinción de membrana)10,11.
El abordaje principal de estos pacientes comienza con evitar el exceso de inmunosupresión o la exposición repetida a drogas antilinfocitarias. Siempre que sea posible, es útil la reducción o cese del tratamiento inmunosupresor.
En general, el manejo del CCM en el trasplantado es similar al de los pacientes no inmunodeprimidos. El tratamiento es fundamentalmente quirúrgico, con resecciones amplias dada su tendencia a la recidiva local12,13. La radioterapia puede ser un tratamiento adyuvante, constituir el tratamiento paliativo en lesiones irresecables o pacientes inoperables, o indicarse como tratamiento de rescate en enfermedad recurrente8,14. La quimioterapia adyuvante no parece mejorar la supervivencia ni disminuir las recurrencias, por lo que se reserva para el tratamiento de la enfermedad de alto riesgo8,15.
En el curso de la enfermedad se presenta afectación ganglionar locorregional en el 55–75%11,16,17, metástasis a distancia (ganglio linfático, piel, hígado, huesos, pulmones y sistema nervioso central) en el 33%11,13 y recurrencias en el 25 y el 44% de los casos13,14.
Cuando el tumor está limitado a la piel, la supervivencia a los 5 años puede alcanzar el 81% en algunas series; mientras que disminuye al 11% en casos de enfermedad diseminada. De igual modo, el pronóstico es peor cuando el tumor afecta a inmunodeprimidos, en donde la supervivencia es del 44% a los dos años, frente al 65–75% de supervivencia cuando se trata de personas no inmunocomprometidas18.
Finalmente, y atendiendo tanto a la mayor incidencia del CCM en pacientes inmunocomprometidos como a las mayores tasas de recidiva, a la afectación metastásica o a la elevada mortalidad a pesar de tratamiento adecuado, no parece necesario insistir en la importancia de la precocidad del diagnóstico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.