


Los estudios proteómicos están adquiriendo en los últimos años una gran relevancia, fundamentalmente en lo que hace referencia a su aplicación a la patología humana. Con este fin se están realizando un gran número de estudios en plasma humano, tejidos y diversos líquidos biológicos. La utilidad práctica de los resultados obtenidos con la proteómica en relación con la salud es muy importante. El descubrimiento de marcadores proteicos de afecciones como las cardiovasculares, neurológicas, oncológicas, metabólicas, entre otras, tiene una aplicación clínica inmediata en el diagnóstico, el seguimiento y el tratamiento de estas enfermedades. Se revisa la metodología utilizada actualmente en los estudios proteómicos.
© 2008 SEDYT. Publicado por Elsevier España, S.L. Todos los derechos reservados.
In recent years, proteomic studies have gained a high profile, mainly those related to its application to human pathology. To this end, a large number of studies are being conducted in human plasma, tissues and various body fluids. The practical usefulness of the results of proteomics in health is of great importance. The discovery of protein markers of diseases such as cardiovascular, neurological, oncologic, and metabolic diseases, among others, has immediate clinical application in their diagnosis, monitoring and treatment. The present article reviews the methodology currently used in proteomics studies.
© 2008 SEDYT. Published by Elsevier España, S.L. All rights reserved.
La gran cantidad de información generada por los proyectos de secuenciación de genomas (Proyecto Genoma Humano)1 y la necesidad de descifrar toda esta información han dado lugar a nuevas acciones en el campo de la biología molecular que han conducido al estudio directo de las proteínas y de los valores de expresión del ARN, en lo que se ha denominado era posgenómica.
Precisamente, el término proteómica se utilizó por vez primera en 1995 para indicar la expresión de una parte variable del genoma2, es decir, el conjunto de proteínas que son causantes de las funciones celulares en cada momento (proteoma). Afirmar que el proteoma consta de todas las proteínas presentes en una célula, organismo o líquido biológico en un momento determinado, incluye no sólo las proteínas traducidas, sino también todas las proteínas modificadas por el corte y empalme alternativo de los tránscritos primarios y el procesamiento posterior a la traducción o la combinación de ambos3. El hecho de que un mismo gen pueda dar lugar a diferentes formas proteicas y estas, a su vez, puedan interaccionar con otras proteínas formando complejos proteicos, o que las proteínas presenten distintas modificaciones postraduccionales dando lugar a diversas formas moleculares que puedan estar presentes simultáneamente, hace que el proteoma represente un nivel de complejidad superior al del genoma. Existe, pues, un único genoma pero múltiples proteomas.
Aunque el Proyecto Genoma Humano ya ha identificado un gran número de genes humanos, la información que existe sobre el proteoma humano es todavía escasa. Se hace, por consiguiente, necesario ahondar en el estudio de las diferencias de expresión proteica y de las modificaciones postraduccionales entre tejidos procedentes de individuos sanos y pacientes afectados de una determinada enfermedad, para obtener una información valiosa sobre las bases moleculares y fisiológicas de esa afección4.
De hecho, la identificación de nuevas proteínas en tejido enfermo frente a tejido sano es una prueba directa de la regulación proteica asociada al proceso patológico, frente a la medida indirecta que proporciona el análisis de la expresión de ARN mensajero. Esta medida directa podría permitir una identificación y una validación más rápidas y fiables de marcadores moleculares con valor diagnóstico para una afección, y de potenciales dianas terapéuticas hacia las que dirigir nuevos fármacos. La proteómica tiene, por tanto, dos líneas claras de aplicación en la biomedicina: la orientada a la identificación de marcadores de diagnóstico, pronóstico o respuesta a tratamiento, y la dirigida al desarrollo de herramientas que permitan el análisis simultáneo de paneles de proteínas identificados como marcadores proteicos.
A diferencia del genoma, el proteoma varía constantemente según el estado y la naturaleza de la célula. Estas variaciones pueden producirse por una situación patológica, el tratamiento con un fármaco o los cambios ambientales a los que la célula puede estar sometida. Los estudios comparativos del proteoma de organismos complejos requieren que las proteínas presentes en extractos de tejido sean separadas antes de su análisis.
Esta necesidad se hace patente cuando se considera que en una célula humana están presentes del orden de 10.000 proteínas. Además, el número de copias de una proteína particular en una célula dada puede variar hasta nueve órdenes de magnitud, con la consecuencia de que especies proteicas de baja abundancia pueden ser extremadamente difíciles de detectar y de caracterizar.
A esto se suma otro nivel de complejidad por el hecho de que los genes individuales pueden producir diversas formas proteicas, por ejemplo, a través del inicio de traducción alternativo o el splicing diferencial, mientras que las modificaciones postraduccionales, tales como la fosforilación, dan lugar a cambios dinámicos en las proteínas.
Existe una serie de métodos para el estudio simultáneo de un alto número de proteínas, incluidas electroforesis en geles bidimensionales, espectrometría de masas y combinaciones de espectrometría de masas y cromatografía líquida. Por este motivo, los estudios proteómicos pueden realizarse con dos enfoques instrumentales5. Por un lado, los estudios proteómicos que emplean la espectrometría de masas y, por otro, los análisis proteómicos empleando micromatrices.
Análisis proteómicos por espectrometría de masasGran parte del éxito de la proteómica se ha debido al desarrollo de las potentes herramientas de espectrometría de masas, que permiten llevar a cabo con éxito la identificación de las proteínas y sus modificaciones postraduccionales. No obstante, estos avances han estado asociados a los desarrollos de nuevas tecnologías de separación de mezclas de proteínas: la electroforesis bidimensional, especialmente en el caso de la tecnología DIGE (Differential In Gel Electrophoresis), y la cromatografía líquida multidimensional, que permite separar los péptidos digeridos de una mezcla proteica6,7.
Separaciones previas Electroforesis bidimensional y tecnología DIGELa electroforesis bidimensional sigue siendo la técnica más resolutiva y empleada en los análisis proteómicos, ya que permite aislar las proteínas mediante una doble separación en un gel 2D-PAGE en base al punto isoeléctrico (pI) y al peso molecular (PM). No obstante, sus limitaciones han provocado el desarrollo de una técnica alternativa como es la tecnología DIGE (Differential In Gel Electrophoresis) comercializada por GE Bio-Sciences, que representa, hoy día, un claro avance en el análisis comparativo de la expresión diferencial de proteínas. Esta técnica aventaja a la electroforesis 2D en que minimiza la variabilidad de los geles y disminuye el tiempo de análisis, y permite una cuantificación muy precisa del perfil de expresión.
Cromatografía líquida multidimensionalEsta técnica novedosa, previa digestión de la mezcla compleja de proteínas, es capaz de separar los péptidos resultantes para la posterior identificación de las proteínas. De hecho, permite construir mapas bidimensionales de elución empleando sistemas cromatográficos multidimensionales conectados a un microcolector de fracciones.
Espectrometría de masasLa espectrometría de masas es una tecnología analítica esencial en el contexto de la proteómica debido a su alta capacidad de análisis, su sensibilidad y su precisión en la determinación de las masas moleculares de péptidos y proteínas, así como los espectros de fragmentación que proporcionan información de secuencia8,9. Se puede realizar con:
- Un espectrómetro de masas consistente en una fuente de ionización Matrix Assisted Laser Desorption/Ionization (MALDI) y dos analizadores de tiempos de vuelo en tándem separados por una cámara de colisión, como los utilizados por el sistema 4700 Proteomics Analyzer.
- Un espectrómetro con fuente de ionización de nanoSpray y analizadores híbridos cuadrupolo-tiempo de vuelo (Q-TOF) como el QSTSAR XL. El sistema cromatográfico conectado a la fuente de ionización puede ser un nano HPLC Ultimate de LC Packings idéntico al descrito anteriormente, con capacidad de realizar cromatografía multidimensional.
A título informativo, y en referencia a la primera de estas técnicas, la conocida como espectrometría de masas MALDI-TOF, es preciso decir que responde al hallazgo, relativamente reciente, de que si los fragmentos de las moléculas analizadas se mezclan con una matriz de naturaleza orgánica, la luz del láser resulta absorbida de forma más eficiente y los espectros tienen mayor intensidad con apenas fragmentación10,11.
Este método MALDI-MS resulta optimizado cuando se asocia con un analizador de tiempo de vuelo (TOF), que permite la determinación de la masa en una región de alto vacío se realice mediante una medida muy precisa del período desde la aceleración de los iones en la fuente hasta que impactan con el detector.
El hecho de que la ionización por MALDI permita detectar moléculas termolábiles, como son las proteínas de forma intacta, la incluye dentro de los métodos de ionización suave (soft-ionization). No obstante, durante el proceso de aceleración o durante el vuelo a través del tubo, se da un proceso de descomposición metastable denominado PSD (post source decay). El análisis de los iones que se producen mediante este fenómeno proporciona una información estructural de la molécula original muy útil, si bien se hace necesario separar estos fragmentos. En un analizador de tipo lineal esto no es posible, ya que los iones formados por PSD tienen la misma velocidad que el ion original y viajan juntos hasta el detector. En la separación se emplean analizadores de tipo reflector que trabajan con voltajes variables para tantos espectros como voltajes empleados que, finalmente, se pegan con ayuda informática para obtener un único espectro de fragmentación11.
Sistemática y fundamentos de la separación e identificación avanzada de proteínasLa identificación de proteínas se realiza tanto de muestras en solución, como de proteínas aisladas en gel de poliacrilamida. Todas las identificaciones incluyen la escisión automática de las bandas de geles bidimensionales y la digestión tríptica de la muestra.
Como plataformas robóticas para el procesamiento de muestras de forma automatizada se pueden utilizar los sistemas Investigador de Genomic Solutions, con capacidad de procesado de 768 muestras simultáneas:
- Investigator ProPic® para la escisión de fragmentos de gel de poliacrilamida, que dispone de dos transiluminadores, de luz visible y UV, para usar según el tipo de gel.
- Investigator ProPrep® para digestión de proteínas en los fragmentos de poliacrilamida, y para la preparación de las muestras para su análisis por huella peptídico asegurando la trazabilidad y la ausencia de contaminación en las muestras.
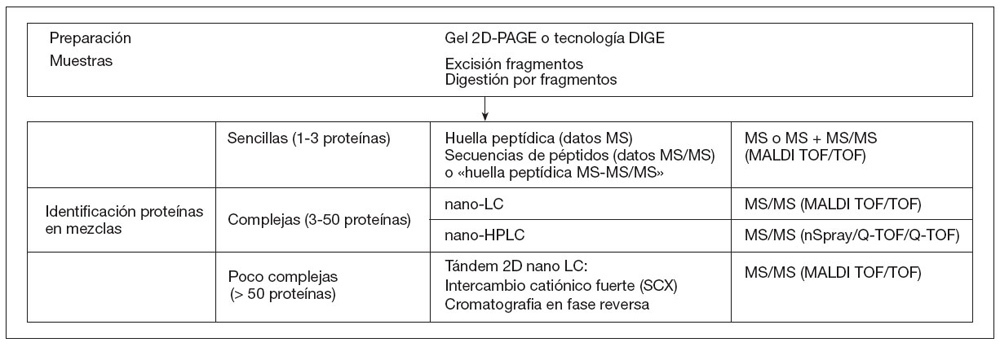
El procesado de las muestras se lleva a cabo de modo diferente atendiendo a su complejidad:
En la figura 1 se recoge esquemáticamente resumidas las tecnologías utilizadas en los análisis proteómicos.
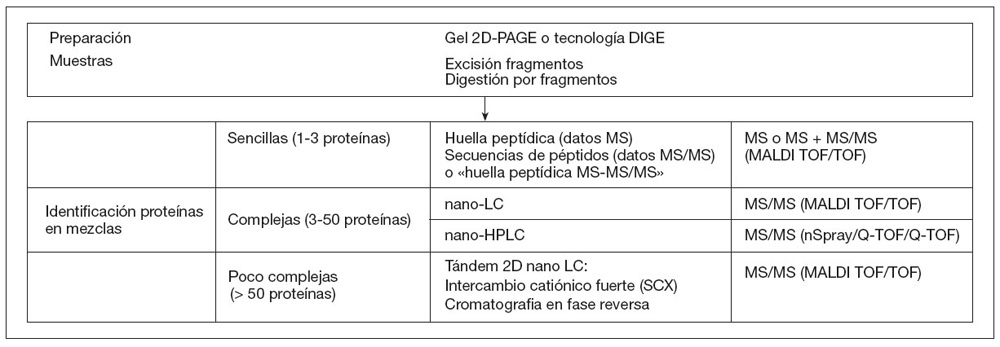
Figura 1. Diagrama esquemático de las tecnologías utilizadas en la técnicas proteómicas. Técnicas para el análisis simultáneo de paneles de proteínas (microarrays)
La genómica funcional y, en concreto, la proteómica están facilitando la identificación de nuevas proteínas y su asociación a enfermedades complejas, como el cáncer o que transcurren a través de procesos inflamatorios como la artritis reumatoide, la psoriasis o la enfermedad inflamatoria intestinal. Decenas de estas proteínas se han convertido en marcadores tanto del diagnóstico, como del pronóstico o la respuesta al tratamiento. Es por ello que resulta necesario el desarrollo de herramientas que permitan el análisis simultáneo de decenas o incluso centenas de proteínas.
La tecnología de los microarrays permite la investigación de cientos de parámetros en un solo experimento. Gracias a esta tecnología, moléculas de interés son inmovilizadas de forma ordenada en soportes sólidos para posteriormente competir con otras moléculas marcadas con el fin de detectar interacciones específicas. Esta interacción puede medirse gracias a sistemas de escaneado y lectura. El desarrollo de los microarrays de proteínas ofrece un importante avance en la miniaturización y automatización del análisis del proteoma, así como en la reducción de costes asociado al bajo consumo de reactivos.
Las aplicaciones de los arrays de proteínas son múltiples, como por ejemplo, el cribado de interacciones proteína-proteína, la identificación de sustratos para proteincinasas y la identificación de proteínas diana para small molecules. En general, este tipo de aplicación puede revolucionar tanto el campo del conocimiento, del área clínica (diagnóstico, monitorización del paciente durante el tratamiento, etc.), como del área farmacéutica (búsqueda de nuevas drogas)4,7.
Existen diferentes tipos de moléculas de captura (capture molecule) depositadas en el array. Estas moléculas pueden ser péptidos, anticuerpos, proteínas, fragmentos de proteínas, aptámeros, enzimas, sustratos, compuestos de bajo peso molecular y scaffolds.
Durante el proceso de la hibridación, el array es expuesto a una mezcla compleja de proteínas, anticuerpos, etc., dependiente del modelo de array elegido. Las uniones específicas pueden entonces monitorizarse siguiendo diferentes aproximaciones basadas en métodos de detección por fluorescencia, quimioluminiscencia, resonancia superficial del plasmón, radiactividad y espectrometría. La detección basada en fluorescencia (con moléculas como Cy3 y Cy5) está ampliamente extendida. La misma instrumentación que se usa para la lectura de los arrays de ADN es aplicable a los arrays de proteínas. Asimismo, existen sistemas para la amplificación de la señal fluorescente entre 10 y 100 veces. La sensibilidad puede pontenciarse con la utilización de métodos fluorescentes y sistemas como la tecnología planar waveguide (Zeptosens), la tecnología basada en beads y partículas en suspensión usando ficoeritrina como marcaje (Luminex®) o la basada en las propiedades de semiconductor de nanocristales (Quantum Dot®).
El progreso de los arrays de proteínas, tanto para investigación como para aplicaciones clínicas, ha sido o está siendo más lento que el de los arrays de ADN debido a las características propias de las moléculas de proteínas, muy distintas de las del ADN. El ADN es una molécula muy uniforme compuesta por 4 nucleótidos, con una estructura hidrofílica bien definida. Por el contrario, las proteínas son moléculas muy diversas construidas a partir de 20 diferentes aminoácidos; pueden ser hidrofílicas o hidrofóbicas, ácidas o básicas. Adicionalmente, debido a la complementariedad del ADN, la interacción específica entre moléculas de ADN puede predecirse de forma fácil. En el caso de las proteínas, debido a la diversa y singular estructura de cada proteína, la interacción entre proteínas no puede predecirse en función de su secuencia de aminoácidos. Glucosilaciones, acetilaciones o fosforilaciones aumentan la diversidad de cada proteína al mismo tiempo que influyen en su interacción con otras proteínas. Por último, resulta relativamente fácil generar las moléculas de ADN para depositar en el array, ya sean productos de PCR u oligonucleótidos sintéticos.
Pero no existe un sistema equivalente para el caso de las proteínas. A pesar de todo ello, los arrays de proteínas constituyen la progresión natural de los ampliamente extendidos y aceptados arrays de ADN. El gran potencial que ofrecen los arrays de proteínas empuja a su estudio y desarrollo. La mejora de la técnica y la puesta a punto de esta tecnología son fundamentales para obtener el máximo beneficio de esta prometedora herramienta.
Biomarcadores proteicos en clínicaUno de los problemas más importantes de la biomedicina es que muchas enfermedades se diagnostican en una etapa avanzada de su desarrollo, lo que dificulta o incluso imposibilita su tratamiento y curación. Por esto hay que disponer de métodos que permitan establecer de manera temprana la disposición a presentar una determinada enfermedad.
En general, las enfermedades no producen alteraciones de una única proteína, sino de muchas proteínas celulares y, especialmente, los procesos agudos producen variaciones de los procesos de modificación de las proteínas posteriores a la traducción. Los procesos crónicos, además de estas alteraciones de los procesos de modificación de las proteínas posteriores a la traducción, producen también cambios de la expresión de los genes y, como consecuencia, variaciones de las concentraciones de proteínas celulares.
Los primeros estudios proteómicos, aunque en esa época no se utilizaba este término, se realizaron en los años ochenta. La puesta a punto de métodos de electroforesis bidimensional en gel, de acuerdo con la técnica de OFarrell12 condujo al análisis de las proteínas de diversos tipos celulares, orgánulos subcelulares y plasma sanguíneo en condiciones normales y en diversas situaciones, tanto fisiológicas como patológicas13-15. Los objetivos de estos estudios eran la identificación del mayor número de proteínas de una célula o de un tejido y la elaboración de listados proteicos. Más adelante, los estudios proteómicos se han aplicado también al análisis de las interacciones entre las proteínas en el contexto de su acción bioquímica.
Como se ha venido señalando en las descripciones anteriores, se utilizan potentes bases de datos y programas informáticos, de forma que se superpongan las masas teóricas de los péptidos con las obtenidas experimentalmente con el fracionamiento de las proteínas16-18. Con los datos de los péptidos que proporciona el espectrómetro de masas, la proteína que se estudia se obtiene utilizando las denominadas huellas de masa de los péptidos o a través de los iones producto.
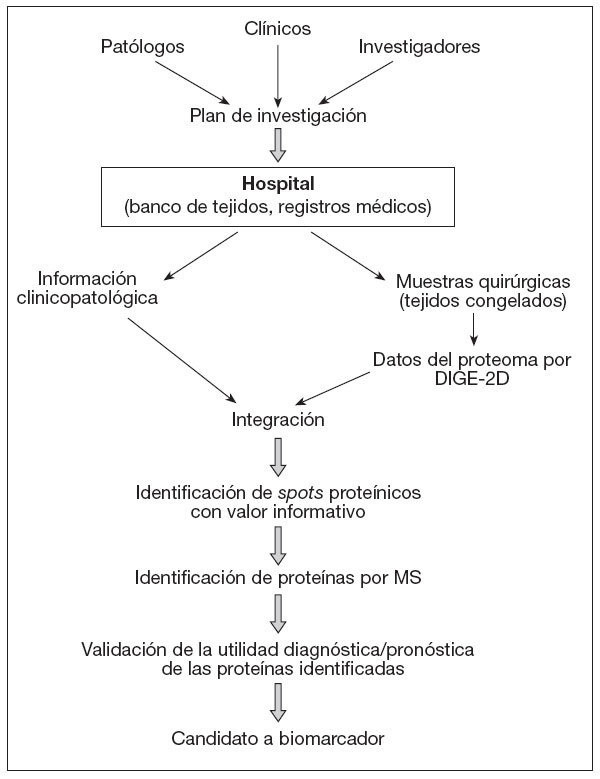
En la figura 2, y a modo de ejemplo, se ilustra la sistemática que se debe seguir para el desarrollo de biomarcadores en estudios de proteómica de cáncer. La tecnología DIGE bidimensional genera los datos del proteoma para las muestras clínicas, que después son examinados en relación con la información clinicopatológica de los donantes. Las manchas o spots proteínicos con valor informativo se someten a identificación utilizando tratamiento de datos por Data Mining, y las proteínas correspondientes son determinadas por espectrometría de masas. En subsiguientes estudios de validación se utilizan los anticuerpos específicos frente a las proteínas identificadas, que también serán utilizadas como parte del examen clínico.
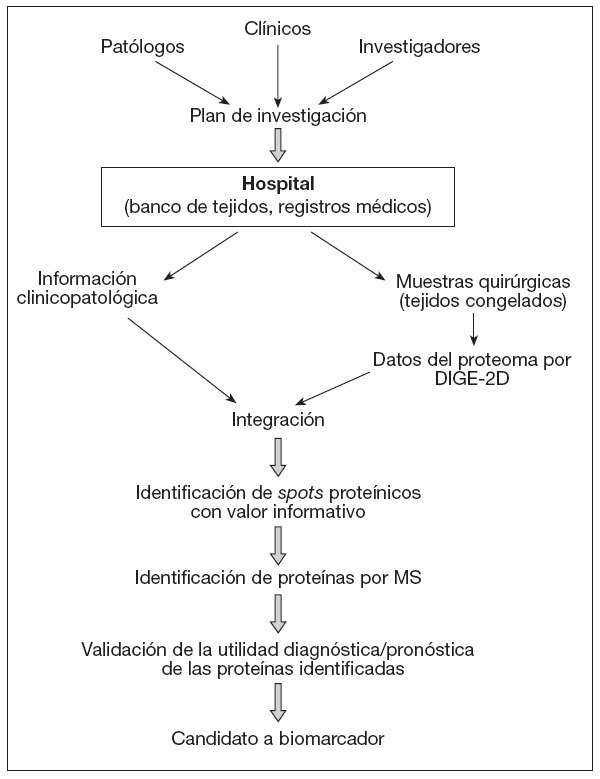
Figura 2. Diagrama de flujo en estudios de proteómica de cáncer para desarrollo de biomarcadores. Conclusiones
La Organización del Proteoma Humano (Human Proteome Organization [HUPO], www.hupo.org) se creó en febrero de 2001 para impulsar un mayor conocimiento de la importancia de la proteómica y las oportunidades que ofrece en el diagnóstico, pronóstico y tratamiento de las enfermedades. Se han constituido varios grupos: HPPP (Human Plasma Proteome Project), HLPP (Human Liver Proteome Project), PSI (Proteome Standards Initiative), HBPP (Human Brain Proteome Project) y MRPP (Mouse and Rat Proteome Project).
Uno de los objetivos de la proteómica es la identificación de marcadores de enfermedad. Un planteamiento ha sido comparar la expresión proteica de los tejidos normales y enfermos para identificar proteínas que se expresen de forma aberrante que puedan representar nuevos marcadores. Otra estrategia es el análisis de las proteínas segregadas en líneas celulares y cultivos primarios y finalmente la obtención de perfiles proteicos en suero.
La proteómica se está aplicando en muchos campos de la patología humana. Entre ellos, las enfermedades cardiovasculares19,20, las neurológicas21, el cáncer22-24 y las enfermedades metabólicas25 y autoinmunitarias26,27.
Declaración de conflicto de interesesLos autores declaran que no existe conflicto de intereses.
INFORMACIÓN DEL ARTÍCULOHistoria del artículo:
Recibido el 23 de diciembre de 2008
Aceptado el 31 de octubre de 2009
* Autor para correspondencia.
Correo electrónico: asanmiguel@hurh.sacyl.es (A. San Miguel-Hernández).