


El diseño de fármacos asistido por computadora (DIFAC) tiene como objetivos el diseño, optimización y selección de compuestos con actividad biológica. El DIFAC forma parte de un esfuerzo multidisciplinario y tiene numerosas aplicaciones específicas durante el proceso de desarrollo de fármacos. A la fecha ha tenido contribuciones significativas en el diseño de fármacos que se encuentran en uso clínico. Es por esto que DIFAC cobra cada vez mayor importancia en la investigación que se hace en la industria farmacéutica, en universidades y centros de investigación. Métodos empleados en DIFAC pueden aplicarse a otras áreas, por ejemplo, productos naturales, bioquímica, química en alimentos, orgánica y teórica. En este artículo se discuten ejemplos de proyectos de diseño de fármacos realizados por un grupo de investigación enfocado en el DIFAC.
The goals of computer-aided drug design (CADD) are the design, optimization and selection of compounds with biological activity. CADD is part of a multidisciplinary effort and has several specific applications during the drug development process. So far, this discipline has made significant contributions to the development of drugs that are currently in clinical use. Therefore, CADD has an increasing relevance in the research performed at the pharmaceutical industry, universities and research centers. Methods used in CADD can be used in other research areas such as natural products, biochemistry, food, organic, and theoretical chemistry. Herein, we discuss examples of drug design projects performed by an academic group focused on CADD.
El diseño de fármacos asistido por computadora (DIFAC) cobra cada vez mayor importancia en la investigación y desarrollo de medicamentos. Esto se ha favorecido por el número de aplicaciones exitosas de métodos de cómputo para el desarrollo de compuestos que actualmente se encuentran en el mercado (Medina-Franco, Lopez-Vallejo y Castillo, 2006). Así mismo, diversos métodos que se emplean frecuentemente en DIFAC pueden transferirse a otras áreas del conocimiento de la química.
El DIFAC forma parte de un esfuerzo multidisciplinario y está conformado por una serie de disciplinas científicas que abarcan modelado molecular, quimioinformática, química teórica y química computacional. A pesar de los avances en el desarrollo de programas computacionales que son de fácil acceso y uso, la aplicación adecuada de las técnicas no es trivial y se debe evitar la idea de «diseñar fármacos apretando botones». La práctica incorrecta de usar métodos de cómputo como cajas negras contribuye a crear falsas expectativas de que los métodos de cómputo pueden diseñar fármacos por sí solos. Esta falsa percepción del DIFAC conduce a preguntas como: «¿Algún medicamento se ha diseñado por métodos computacionales?» Efectivamente, los métodos de cómputo no diseñan de forma automatizada los medicamentos. Los modelos computacionales deben estar integrados con pruebas experimentales, usualmente por medio de varios ciclos de optimización. Si bien es cierto que el procesamiento de datos puede ser, en general, rápido, la elección de métodos, el análisis e interpretación de datos son laboriosos. Al igual que las pruebas experimentales, las metodologías computacionales deben validarse para encontrar los parámetros óptimos que den resultados confiables.
Diversas universidades y centros de investigación en el mundo cuentan con grupos especializados en el desarrollo y/o aplicación de metodologías computacionales para la investigación de fármacos. Algunas universidades ya incluyen en sus planes de estudio el tema de DIFAC. Por ejemplo, se inició el grupo de Diseño de Fármacos Asistido por Computadora de la Facultad de Química (DIFACQUIM) de la Universidad Nacional Autónoma de México. DIFACQUIM trabaja en un ambiente multidisciplinario colaborando con grupos en México, Estados Unidos, varios países en Europa y Asia.
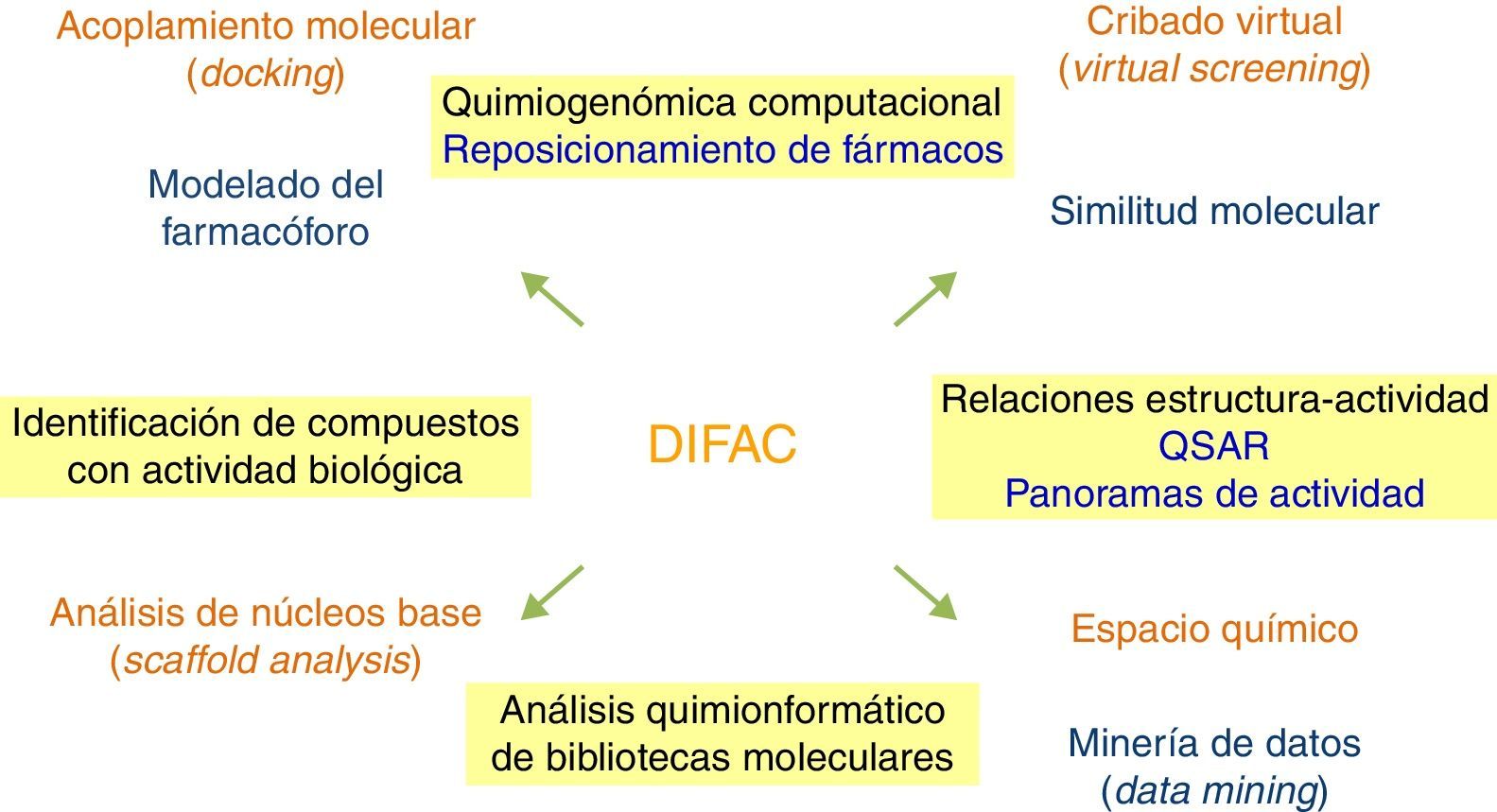
La figura 1 ilustra líneas de investigación que típicamente pueden seguirse en un grupo académico enfocado en el DIFAC. La figura también muestra conceptos y métodos representativos que se emplean dependiendo de los objetivos particulares de cada proyecto. En este artículo se discuten estas líneas de investigación representativas describiendo los objetivos principales y los métodos empleados con frecuencia. Al final se discuten brevemente 2 casos de estudio.
Modelado molecular de compuestos con actividad biológica
Entender el modo de acción de moléculas activas a nivel molecular tiene gran interés científico y práctico. Una de las aplicaciones es proponer cambios específicos a las estructuras químicas para ayudar a incrementar su afinidad con la diana terapéutica y, en principio, aumentar su actividad biológica. Típicamente, los objetivos de esta línea de investigación son 3: 1) encontrar nuevos compuestos líder para su posterior optimización; 2) identificar compuestos selectivos; y 3) optimizar la actividad biológica de compuestos activos. Para alcanzar estos objetivos, y dependiendo de la información experimental disponible se emplea con frecuencia acoplamiento molecular automatizado (en inglés molecular docking), cribado o filtrado computacional de colecciones de compuestos (virtual screening) y modelado del farmacóforo (pharmacophore modeling).
El acoplamiento molecular automatizado tiene como finalidad buscar la conformación y posición óptima de un ligando dentro de un blanco molecular o la posición y conformación más favorable de 2 macromoléculas (Bello, Martinez-Archundia y Correa-Basurto, 2013). El cribado virtual consiste en filtrar series de compuestos, normalmente bases de datos moleculares grandes, para seleccionar un subconjunto de moléculas que se sometan a ensayos biológicos (Lavecchia y Di Giovanni, 2013). El filtrado se hace empleando uno o más métodos computacionales tales como acoplamiento molecular o similitud molecular. El cribado virtual debe estar integrado con la validación experimental y normalmente se hace en forma de 2 o más ciclos de refinamiento de los resultados. Por su parte, el modelado del farmacóforo tiene como finalidad detectar en forma automatizada el arreglo tridimensional de las características mínimas necesarias estéricas y electrónicas para asegurar interacciones óptimas con un blanco farmacológico específico (Sanders et al., 2012). Se desea que estas características desencadenen la respuesta biológica esperada. En DIFACQUIM se emplean estas metodologías para el desarrollo de inhibidores de ADN metiltransferasas (DNMT, por sus siglas en inglés) (vide infra).
Quimiogenómica computacional y reposicionamiento de fármacosLa quimiogenómica está enfocada a conocer las asociaciones que hay entre compuestos químicos y dianas moleculares (Rognan, 2010). Algunos objetivos representativos de esta área son: 1) identificar compuestos activos en bases de datos moleculares; 2) encontrar blancos moleculares para compuestos activos; y 3) proponer usos terapéuticos alternos para fármacos aprobados (reposicionamiento de fármacos, vide infra). La quimiogenómica está asociada al concepto de polifarmacología (Rosini, 2014) que consiste en la interacción «favorable» de un fármaco con una serie de dianas biológicas para tener un efecto clínico esperado. Desde luego, un fármaco puede presentar múltiples interacciones con dianas terapéuticas que están asociadas con efectos adversos. Así, uno de los principales retos del diseño de fármacos actual es la identificación de «llaves maestras»: fármacos que interaccionen con dianas biológicas deseadas pero que no interaccionen con blancos moleculares asociados con efectos adversos (off targets) (Medina-Franco, Giulianotti, Welmaker y Houghten 2013).
Ejemplos de conceptos y métodos computacionales que se emplean con frecuencia en quimiogenómica computacional son el cribado virtual y búsqueda computacional de blancos moleculares (target fishing). Este último puede entenderse como el proceso complementario al filtrado computacional: identificación de uno o varios blancos moleculares de compuestos activos. En quimiogenómica también se emplean con frecuencia métodos quimioinformáticos (vide infra) y de similitud molecular. Similitud molecular es un concepto abstracto por el cual trata de cuantificarse el parecido que hay entre 2 estructuras químicas. La similitud molecular depende altamente de la representación molecular y de la métrica para cuantificar el «grado de acercamiento» de las representaciones moleculares (Medina-Franco y Maggiora, 2014).
Relaciones estructura-actividad y estructura-múltiple actividadEstablecer relaciones estructura-propiedad (structure-property relationships [SPR]) es una práctica común en muchas áreas de la química. En farmacia es frecuente enfocarse a la actividad biológica como la propiedad de interés. De esta manera, los estudios de relaciones estructura-actividad (structure-activity relationhips [SAR]) son muy comunes en desarrollo de fármacos. Recientemente, y debido al interés creciente de la comunidad científica en polifarmacología, es recurrente que series de compuestos se evalúen con 2 o más blancos biológicos. Así, los estudios SAR pueden extenderse a estudios de relaciones estructura múltiple-actividad (structure-multiple activity relationships [SmAR]). Estudios SAR/SmAR que se efectúan en DIFAC tienen como objetivos: 1) optimización de la actividad biológica de compuestos activos; 2) desarrollo de métodos para análisis cuantitativo y visual del SAR; 3) análisis SAR/SmAR de series de compuestos; y 4) identificación de activity cliffs en forma sistemática.
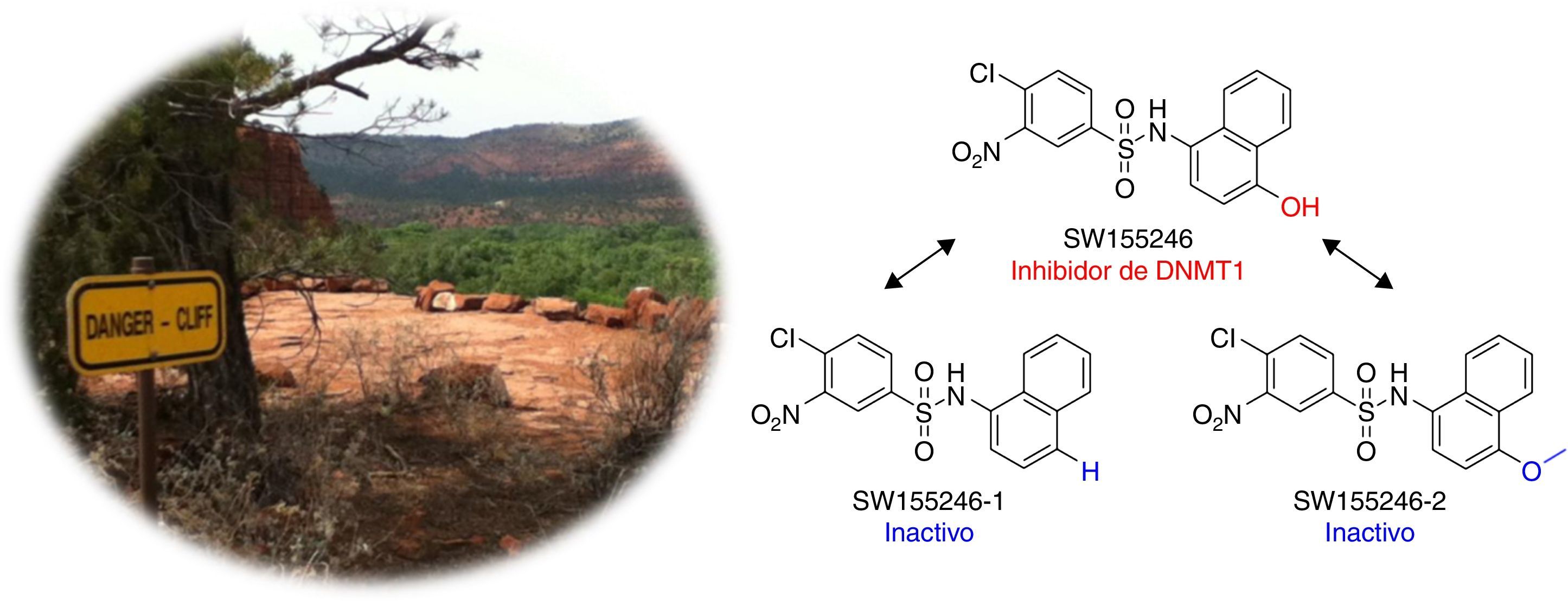
Dos ejemplos representativos de conceptos que se emplean en este tipo de estudios son Quantitative Structure-Activity Relationships (QSAR) y panoramas de actividad o panoramas de propiedad (activity/property landscapes). En QSAR se busca encontrar relaciones cuantitativas estructura-actividad (Tropsha, 2010). Este tipo de métodos asumen que moléculas semejantes tienen propiedades semejantes. Sin embargo, hay casos de compuestos que tienen estructuras químicas muy parecidas, pero que tienen actividades muy diferentes. A estos casos se les denomina acantilados (o pozos) de actividad (activity cliffs) (Maggiora, 2006). Un ejemplo se ilustra en la figura 2.
 que se encuentra en la naturaleza (estado de Arizona, EE. UU.).")
Los acantilados o pozos de actividad (donde la «profundidad» depende de la magnitud de la diferencia de actividad) tienen 2 facetas opuestas. Por una parte, los acantilados de actividad dan información farmacofórica que puede ser de gran utilidad en programas de optimización. Por el contrario, pueden tener efectos negativos en el desarrollo de modelos como QSAR; la presencia de acantilados de actividad hace que estos modelos sean poco predictivos (Cruz-Monteagudo et al., 2014).
En grupos de investigación académica como DIFACQUIM se han realizado estudios enfocados a explorar los panoramas de actividad de moléculas evaluadas contra uno o varios blancos biológicos (Medina-Franco y Waddell, 2012), incluyendo el análisis SmAR de compuestos obtenidos por química combinatoria. Estos estudios se han extendido al estudio de SPR de factores relacionados con la absorción, distribución metabolismo y excreción de fármacos (Rojas-Aguirre y Medina-Franco, 2014). Los principios para el estudio de SAR/SPR se han aplicado al análisis cuantitativo de relaciones estructura de ligandos con las interacciones ligando-proteína calculadas a partir de complejos tridimensionales (Méndez-Lucio, Kooistra, Graaf, Bender y Medina-Franco, 2015).
Análisis quimioinformático de bases de datos molecularesLas bases de datos de compuestos, incluyendo las que contienen información de actividad biológica, forman parte muy importante del descubrimiento y desarrollo de fármacos. Estas bases de datos pueden contener información de hasta millones de estructuras con datos de actividad biológica y son parte de lo que se denomina big data (Lusher, McGuire, van Schaik, Nicholson y de Vlieg, 2014). De esta manera, la minería eficiente de datos de estas colecciones requiere de métodos informáticos que forman parte de lo que se llama «quimioinformática» (Duffy, Zhu, Decornez y Kitchen, 2012). Hay diversas definiciones de este campo del conocimiento, pero todas ellas tienen en común el manejo de información química con métodos de cómputo. La quimioinformática tiene diversas aplicaciones no solamente en farmacia, sino también en otras áreas como química analítica y orgánica. Respecto al manejo de bases de datos, la quimioinformática se emplea para analizar cuantitativamente la diversidad química, la visualización del espacio químico y el contenido y diversidad de núcleos base (molecular scaffolds), entre otras aplicaciones.
Es común que en grupos dedicados al DIFAC se realicen análisis quimioinformáticos de bases de datos moleculares para obtener su perfil en al menos 3 aspectos: propiedades fisicoquímicas, diversidad estructural y distribución en el espacio químico. Ejemplos de estos análisis son la caracterización de colecciones de productos naturales (Yongye, Waddell y Medina-Franco, 2012), compuestos obtenidos por química sintética y fármacos aprobados (López-Vallejo, Giulianotti, Houghten y Medina-Franco, 2012).
Caso de estudio: desarrollo de inhibidores de ADN metiltransferasasEl desarrollo asistido por computadora de inhibidores de DNMT ilustra la aplicación de diversas técnicas empleadas en DIFAC dirigidas a resolver problemas de salud pública.
La metilación de ADN es un proceso epigenético que se lleva a cabo en la posición C5 de residuos de citosina. El proceso es catalizado por la familia de enzimas DNMTs. A esta familia pertenecen DNMT1, DNMT3A, DNMT3B y DNMT3L. DNMT1 es la más abundante y está relacionada con el mantenimiento del patrón de metilación del ADN durante la replicación celular en mamíferos. DNMT3A y DNMT3B, asociadas con DNMT3L, actúan como metilantes de novo del ADN (Robertson, 2001). El mecanismo de metilación no se conoce con certeza. Sin embargo, los aminoácidos que se proponen que participan en el mecanismo han guiado estudios de acoplamiento molecular (Medina-Franco, Méndez-Lucio, Yoo y Dueñas, 2015).
Los cambios epigenéticos no modifican los genes, pero regulan su expresión. De esta manera, si existen factores externos que dañen estos mecanismos se pueden provocar enfermedades cardiacas, psiquiátricas o cáncer (Medina-Franco et al., 2015). En especial, se ha identificado que los niveles de expresión de las DNMT son elevados en el cáncer de colon, próstata, mama, hígado y en leucemia, lo que a su vez está vinculado con el silenciamiento de los genes supresores de tumores debido a mecanismos epigenéticos (Subramaniam, Thombre, Dhar y Anant, 2014). De esta manera, se plantea que el diseño de inhibidores de DNMT (iDNMT) es una estrategia para reducir la hipermetilación asociada a distintas enfermedades relacionadas con fenómenos epigenéticos. Hoy en día se encuentran en uso clínico 2 iDNMT, la azacitidina y la decitabina, ambos aprobados por la Food and Drug Administration (FDA) de Estados Unidos para el tratamiento de síndromes mielodisplásicos. Sin embargo, estos fármacos son compuestos nucleosídicos y tienen problemas de citotoxicidad. Es por esto que hay un especial interés para identificar y desarrollar nuevos inhibidores de DNMT que no sean nucleosídicos. A la fecha se conocen diversos iDNMT que se han identificado por métodos diversos y que tienen orígenes diferentes, incluyendo productos naturales, fármacos obtenidos por reposicionamiento, «hits» de cribado virtual y compuestos de origen sintético (Erdmann, Halby, Fahy y Arimondo, 2014). Dentro de las técnicas computacionales más utilizadas en epigenética se encuentran acoplamiento molecular, modelado por homología (Heinke, Carlino, Kannan, Jung y Sippl, 2011), dinámica molecular (Deschamps, Simões-Pires, Carrupt y Nurisso, 2015) y filtrado computacional (Kuck, Singh, Lyko y Medina-Franco 2010).
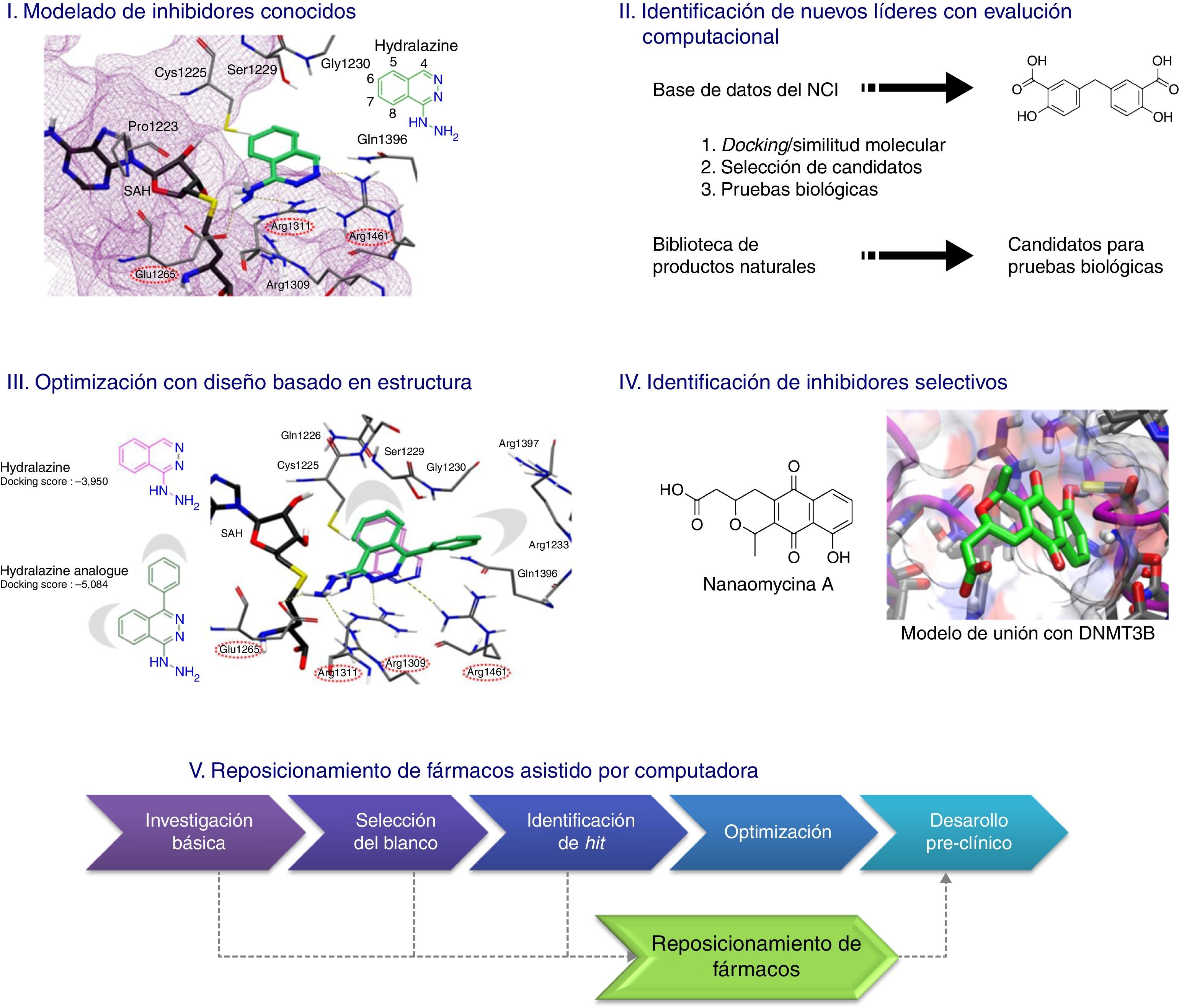
Con la finalidad de desarrollar nuevos iDNMT se han establecido programas de investigación en grupos académicos cuya estrategia general es la aplicación de DIFAC. La investigación se realiza en grupos multidisciplinarios. En DIFACQUIM se tienen 4 objetivos específicos: 1) entender las relaciones estructura-actividad de iDNMT conocidos; 2) identificar nuevos inhibidores o moduladores de DNMT; 3) optimizar iDNMT líderes; y 4) reposicionamiento de fármacos. Para alcanzar estos objetivos se han planteado 5 estrategias computacionales (fig. 3).

I) La primera estrategia es el modelado molecular de inhibidores conocidos. El acoplamiento molecular y modelado del farmacóforo son las metodologías principales que se han empleado a la fecha.
II) La segunda estrategia es el cribado virtual de bases de datos moleculares para la identificación de nuevos iDNMT. El cribado virtual se ha realizado mediante acoplamiento molecular y similitud molecular. A la fecha se ha hecho el filtrado de bases de datos públicas, de productos naturales y compuestos de origen sintético.
III) La tercera estrategia es el diseño de nuevos inhibidores basado en la estructura de complejos ligando-proteína. Aquí, el punto de partida son modelos de acoplamiento que se proponen en la primera estrategia. El objetivo es encontrar las sustituciones específicas que se puedan realizar a los inhibidores para incrementar las interacciones en la diana molecular. Se espera que la mayor afinidad molecular se traduzca en un incremento en la actividad biológica (aunque se sabe que hay otros factores asociados a la actividad biológica que no se consideran en el desarrollo de modelos ligando-proteína).
IV) La cuarta estrategia es la identificación de inhibidores selectivos hacia las diferentes isoformas. No solamente se buscan compuestos que puedan tener un fin terapéutico. También se espera que compuestos altamente selectivos puedan desarrollarse como moléculas sonda epigenéticas (epi-probes) para ensayos bioquímicos. Por ejemplo, en DIFACQUIM se realizan estudios de acoplamiento molecular rígido-flexible y flexible-flexible con el propósito de estudiar cuáles son las interacciones importantes de iDNMT con DNMT3A, inicialmente identificados por filtrado computacional y luego optimizados mediante síntesis (Kabro et al., 2013). Se espera obtener la información necesaria para proponer modificaciones específicas que aumenten la selectividad y potencia.
V) La quinta estrategia (fig. 3) es el reposicionamiento de fármacos asistido por computadora. La finalidad es encontrar fármacos aprobados para otra indicación que sean activos contra DNMT y que puedan tener una nueva indicación como epi-fármacos.
Como parte de los proyectos enfocados al desarrollo de iDNMT que se desarrollan en DIFACQUIM se está generando una base de datos molecular que colecta la información de inhibidores de las distintas isoformas de la DNMT. Esta base de datos molecular sirve como punto de partida para caracterizar el espacio químico relevante de los iDNMT conocido hasta la fecha. La caracterización está enfocada a analizar la diversidad estructural y cobertura del espacio químico.
Caso de estudio: reposicionamiento de fármacos asistido por computadoraEl reposicionamiento de fármacos es un proceso por medio del cual se busca que fármacos dirigidos para tratar una enfermedad puedan usarse para tratar otra distinta (Ashburn y Thor, 2004). Esta estrategia se apoya en hipótesis teóricas y en observaciones de casos reales que demuestran que fármacos aprobados para tratar una enfermedad pueden tener otra indicación diferente. Uno de los pilares en los que se apoya el reposicionamiento es la polifarmacología. Se estima que un fármaco puede interactuar directamente con entre 6 y 13 dianas en promedio (Vogt y Mestres, 2010).
Los primeros casos exitosos de reposicionamiento fueron por serendipia. Subsecuentemente, se implementaron búsquedas en la literatura (por ejemplo, un investigador interesado en encontrar una cura para una enfermedad busca un fármaco que tenga los efectos deseados). En la actualidad, el reposicionamiento se puede realizar con la ayuda de métodos computacionales, por ejemplo con cribado virtual (vide supra).
El reposicionamiento de fármacos ha mostrado ahorrar hasta el 50% de las inversiones de tiempo y dinero para desarrollar la aprobación de un tratamiento (Paul y Lewis-Hall, 2013). Esto permite un beneficio considerable no solo para las empresas farmacéuticas, sino también para quienes padecen enfermedades poco comunes o tropicales (Ekins, Williams, Krasowski y Freundlich, 2011). El reposicionamiento es una estrategia atractiva puesto que un fármaco aprobado requerirá de menos tiempo para probar su seguridad, ahorrando tiempo y recursos que se invierten durante las fases preclínicas y clínicas. También se ha planteado el beneficio de realizar reposicionamiento de fármacos de manera temprana (Temesi et al., 2014). Finalmente, fármacos que no han probado efectividad para una determinada enfermedad pero sí seguridad, son excelentes candidatos para reposicionamiento ya que cuentan con datos de seguridad en humanos (Turner, Bascomb, Maki, Rao y Young, 2011).
Existe una gran cantidad de fármacos que han pasado por el proceso de reposicionamiento. A continuación se proporcionan algunos ejemplos. El sildenafil es un caso típico. Aun cuando falló demostrar efectividad como antihipertensivo, por serendipia se encontró que podía ser una terapia efectiva contra la disfunción eréctil (Boolell, Gepi-Attee, Gingell yAllen, 1996). Por revisión de la literatura se propuso que podría tratar la hipertensión pulmonar (Ghofrani et al., 2002). Un ejemplo de reposicionamiento en México fue desarrollado por médicos del Instituto Nacional de Nutrición «Salvador Zubirán» (Rull, Quibrera, González-Millán y Castañeda, 1969). Con el conocimiento de la fisiopatología de la neuropatía diabética, y una búsqueda basada en la literatura, se propuso que la carbamazepina sería un fármaco plausible para tratar esta enfermedad. Después de que demostrara efectividad, este tratamiento fue pionero en el tratamiento de la neuropatía diabética. Los fármacos actualmente aprobados para tratar la neuropatía diabética (pregabalina y gabapentina) pertenecen al mismo grupo (anticonvulsivantes) y son otro ejemplo de reposicionamiento. Otro ejemplo de un estudio que se realizó con la participación de DIFACQUIM consistió en probar que ribavirina, que es un inhibidor de eIF4E e IMPDH, también inhibe a EZH2. Un estudio computacional mostró que ribavirina tiene una alta similitud estructural con 3-deazaneplanocin A (De la Cruz-Hernandez et al., 2015).
ConclusionesDiversos conceptos y métodos computacionales tienen un papel muy importante en el proceso de desarrollo de fármacos. Dentro de las aplicaciones específicas se encuentran el descubrimiento de principios activos, el diseño y la optimización de moléculas con bioactividad y la selección de compuestos potencialmente bioactivos. DIFAC es una actividad científica que integra y aplica conocimientos de diversos campos del conocimiento tales como química teórica, computacional, modelado molecular y quimioinformática. DIFAC forma parte de un esfuerzo multidisciplinario y debe estar integrado con otros campos del conocimiento que se emplean en química farmacéutica.
PerspectivasEn DIFAC hay muchos retos que quedan por afrontar. Uno de ellos es el desarrollo de algoritmos que permitan captar cada vez con mayor eficiencia y precisión información biológica relevante para el diseño de fármacos. Con el advenimiento de la polifarmacología se hace más evidente la necesidad del diseño asistido por computadora de moléculas que actúen como «llaves maestras» (específicas para una serie de dianas biológicas). También se espera que los métodos computacionales sean percibidos cada vez menos como herramientas técnicas y que se elimine la percepción del DIFAC como «diseño de fármacos apretando botones». Se anticipa que aumentará la vinculación de DIFAC con otras áreas. Un ejemplo es la asociación con química en alimentos en el campo de investigación llamado FoodInformatics (Martinez-Mayorga y Medina-Franco, 2014).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Se agradece el apoyo del Departamento de Farmacia de la Facultad de Química de la UNAM y al proyecto PAIP 5000-9163 (UNAM) otorgado a JLM-F.
La revisión por pares es responsabilidad de la Universidad Nacional Autónoma de México.