



Hutchinson-Gilford progeria syndrome (HGPS) also known as childhood progeria is a rare genetic disease characterized by accelerated aging beginning in early childhood. The phenotypic features of this syndrome are caused by alterations in the lamin A protein, fibrillar component of the nuclear lamina which maintain the structure of the nuclear envelope and participates in organization chromatin. Children with progeria have a point mutation in the LMNA gene which leads to the production of a permanently farnesylated mutant lamin A called progerin, that contribute to premature aging. In normal protein, this farnesyl group is removed, but this step does not take place in progeria and the progerin remain attached to the inner nuclear membrane, causing alterations in nuclear morphology. The use of several drugs that inhibit the farnesylation of progerin has been proposed as a promising therapeutic approach to reverse the adverse effects of progerin synthesis. Some studies have determined that progerin is also produced in normal individuals and increase with age, suggesting that these studies of progeria can shed light on the normal process of aging. In this paper the main aspects of HGPS such signs and symptoms, genetic basis, and how it has driven the discovery of strategies for treating the disease will be discussed.
El síndrome de Hutchinson-Gilford (HGPS), también conocido como progeria infantil es una enfermedad genética rara, caracterizada por un envejecimiento prematuro que comienza tempranamente en la infancia. Las características fenotípicas de este síndrome son causadas por alteraciones en la proteína de la lámina A, componente fibrilar principal que mantiene la estructura del núcleo y participa en la organización de la cromatina. Los niños con progeria tienen una mutación puntual en el gen LMNA originando una forma anormal de la lámina A farnesilada llamada progerina, que contribuye al envejecimiento prematuro. En la proteína normal este grupo farnesilo es removido, pero en la progeria no ocurre este paso, y la progerina permanece unida a la envoltura nuclear interna, causando alteraciones en la morfología y funcionalidad nuclear. El uso de varios fármacos que inhiben la farnesilación de la progerina ha sido propuesto en algunos ensayos terapêuticos prometedores para revertir los efectos nocivos de la síntesis de la progerina. Algunos estudios han determinado que la progerina también es producida en individuos sanos e incrementa con la edad, lo que puede arrojar información sobre el proceso normal de envejecimiento. En este artículo se discutirán los principales aspectos del HGPS así como sus características, síntomas, bases genéticas y como se ha impulsado el descubrimiento y el desarrollo de estrategias para el tratamiento de la enfermedad.
El término progeria proviene del griego pro, “hacia, a favor de” y geron o geras, “viejo” y significa envejecer prematuramente (Sarkar y Shinton, 2001). Aunque existen diferentes síndromes progeroides, el más común es el síndrome de Hutchinson-Gilford, nombrado así en honor a los médicos ingleses Jonathan Hutchinson (1886) y Hastings Gilford (1904). Estos médicos describieron la progeria por primera vez, por lo que en la literatura frecuentemente es abreviada HGPS (Hutchinson-Gilford progeria syndrome) (HGPS, MIM–17667), también denominada progeria infantil. La progeria tiene una incidencia de 1 niño por cada 4 millones de nacimientos (Gordon, et al., 1993–2013). La progeria infantil es una enfermedad genética rara, incurable y fatal caracterizada por un envejecimiento brusco y prematuro tanto en los niños como en niñas, sin distinción de razas, provocando niños con la apariencia de ancianos. Los niños con HGPS tienen un desarrollo fetal normal y nacen con un aspecto saludable. Sin embargo, entre los 18 y 24 meses de edad aproximadamente, comienzan a manifestar muchos rasgos característicos de la vejez prematura. En estos pacientes se producen alteraciones en la integridad del tejido conectivo, componente esencial de varios órganos y tejidos como: hueso, músculo, piel, tejido subcutáneo y vasos sanguíneos. Los signos de la progeria son la falta de crecimiento, ojos saltones, nariz en forma de pico, pérdida de peso y cabello, arrugas y manchas en la piel, rigidez, dislocación de la cadera, ateroesclerosis generalizada, además de enfermedades cardiovasculares y derrames cerebrales. Algunas de las características anteriores son similares a las encontradas en el envejecimiento humano.
En general, el proceso de envejecimiento se produce 5 a 10 veces más rápido que lo habitual en estos pacientes y por ello aparentan mucho mayor edad que la que tienen (Stables y Morley, 1994) (figura 1). La mayoría de los niños con progeria fallecen de enfermedades cardiacas que afectan a millones de adultos con envejecimiento normal (Gerhard-Herman, et al., 2012). En este contexto, la media de edad de fallecimiento en esta patología es alrededor de los 13 años, con un rango que se extiende de los 8 a los 21 años. Estos niños no sufren la enfermedad de Alzheimer, de cataratas ni de los cánceres típicos del envejecimiento. Otros órganos como hígado, riñones, pulmón, sistema digestivo, médula ósea y cerebro no se ven afectados por la enfermedad (Hennekam, 2006). Un aspecto importante es que los niños con progeria no presentan alteraciones neurológicas, son inteligentes, valientes y están llenos de vida y por esta razón su desarrollo cognitivo y emocional no se correlaciona con el envejecimiento fenotípico (Pollex y Hegele, 2004; Merideth, et al., 2008; Gordon, et al., 1993–2013).
.")
Niños con progeria infantil, cuando todavia no cumplían los 10 anos de edad (tomada de Starr y Taggart, 2008).
El fenotipo característico de este síndrome se debe a alteraciones en la lámina nuclear, estructura formada de filamentos intermedios (lámina A, B y C). Las funciones de la lámina nuclear son: mantener la estructura de la envoltura nuclear y la posición de los poros nucleares, servir de anclaje para la cromatina y de soporte para diversas reacciones asociadas a ella, conformar una plataforma estructural que conecta el núcleo al citoesqueleto de la célula, e influir en la actividad de proteínas que regulan la replicación del ADN, transcripción y regulación del ciclo celular. Las deficiencias de estas lámina producen un grupo de enfermedades muy heterogéneas denominadas laminopatías. A nivel molecular, estas deficiencias son causadas por mutaciones en el gen LMNA que codifica para las lámina de tipo A y C (Burke y Stewart, 2006; Worman, 2012). La laminopatía progeroide más ampliamente estudiada es el síndrome de Hutchinson-Gilford o progeria infantil (Mounkes y Stewart, 2004; Young, et al., 2006) de la cual nos vamos a enfocar en este artículo.
Causas de la progeria infantilLa mayoría de los casos con HGPS presentan mutaciones puntuales en el gen autosómico LMNA, que produce una lámina A incorrecta llamada progerina. La forma anormal de pre-lámina A altera la envoltura nuclear y, en consecuencia, las células se dividen mal o no lo hacen. La restauración y renovación de los tejidos no se puede realizar y se produce un envejecimiento muy rápido. Los núcleos de las células de los niños con progeria están deformados, presentan modificaciones estructurales (herniaciones y lóbulos). Molecularmente presentan alteraciones en la organización de la cromatina, que predispone a rupturas de la doble cadena en el ADN y a una senalización inadecuada para su reparación la cual propicia a un arresto en la replicación celular e inducción de la senescencia o muerte celular (Liu, et al., 2006; Baek, et al., 2013).
La progeria infantil es una enfermedad genética rara con un modelo de herencia autosómico dominante. Sin embargo, los casos típicamente son esporádicos y presentan mutaciones de novo, lo que significa que la enfermedad no fue transmitida por sus progenitores (Stables y Morley, 1994).
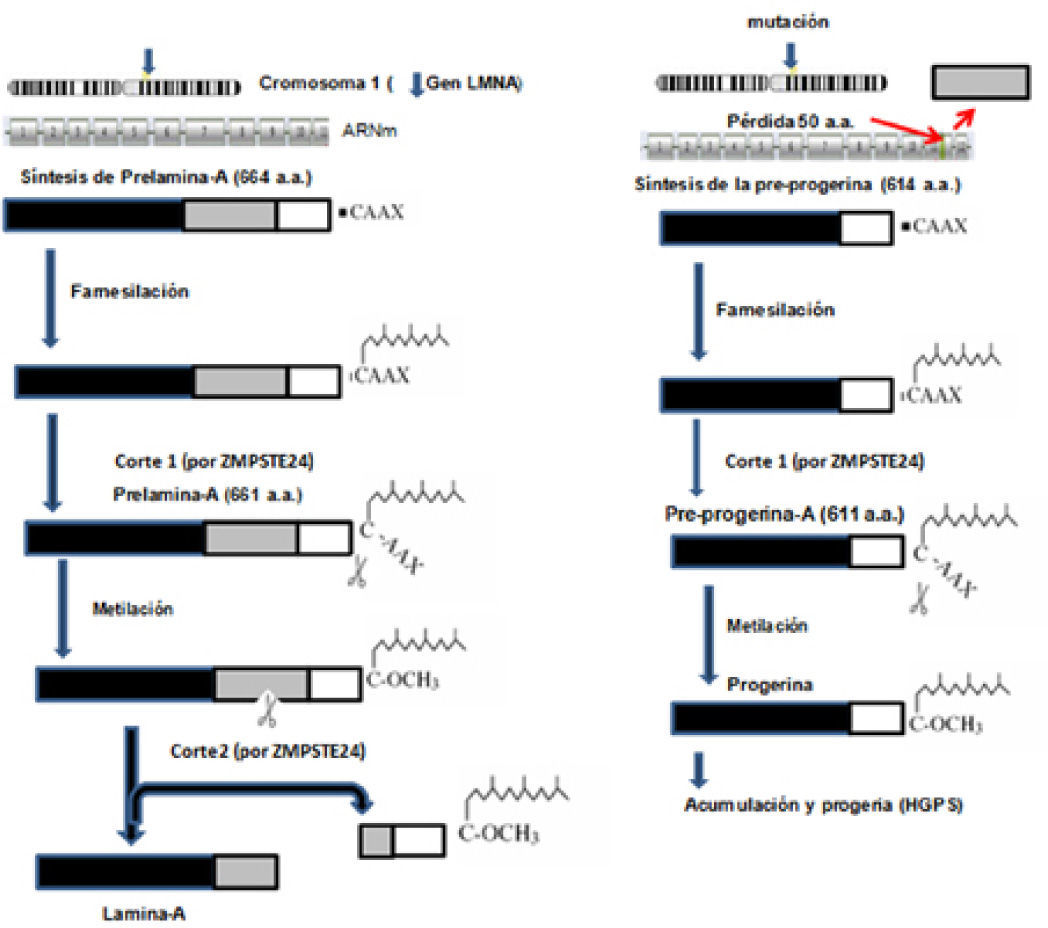
Las lámina A y C son codificadas por el mismo gen (LMNA) que se localiza en el cromosoma 1, mientras que la B es codificada por el gen (LMNB) ubicado en el cromosoma 5. El ARN mensajero (ARNm) que contiene información para la síntesis de la lámina C se forma por la transcripción de LMNA hasta el exón 10, mientras que para la lámina A continúa la transcripción hasta el exón 12. Los dos transcritos resultantes de diferente longitud son traducidos en los ribosomas del citoplasma y originan las lámina: C o A. Por este origen común, la lámina A se considera como una lámina C a la que se le añade un segmento extra de 77 aminoácidos. La lámina A se sintetiza como un precursor denominado prelámina-A que, en su segmento carboxilo-terminal posee 4 aminoácidos (CAAX), donde C es cisteína, A corresponde a un aminoácido alifático y X a cualquier aminoácido. El motivo CAAX induce un proceso ordenado de modificaciones postraduc-cionales (farnesilación, metilación) que serán fundamentales para originar la lámina A madura y correctamente activa. Estos aminoácidos terminales de la lámina A no existen en la lámina C. La mutación más frecuente que causa HGPS es una mutación puntual en la posición 1824 en el exón 11 del gen de la prelámina-A que consiste de la sustitución de una citosina por una timina en la tercera base del codón 608. Esta mutación no cambia el aminoácido codificado, pero activa un punto críptico de corte en el procesamiento del ARNm lo que provoca la síntesis de una variante de prelámina-A con pérdida de 50 aminoácidos en el extremo carboxilo. Esto no afecta el motivo CAAX que es codificado por el exón 12. Esta proteína más corta es conocida como progerina o lámina AD50, que carece de un sitio de corte por la enzima Zmpte24 y que, por lo tanto, afecta específicamente la maduración de la prelámina-A en la lámina A, la cual permanece farnesilada constitutivamente, se acumula en el núcleo y presenta alteraciones estructurales, así como defectos en la organización de la cromatina que desarrollan los síntomas del envejecimiento prematuro (De Sandre-Giovannoli, et al., 2003; Eriksson, et al., 2003).
Síntesis de la lámina A y de la progerinaEl primer paso que sufre la prelámina-A es la farnesilación (unión de un grupo farnesilo, lípido de 15 carbonos), en el grupo tiol de la cisteína (C) del segmento CAAX, por medio de una farnesiltransferasa (FTasa) citosólica. Así, esta proteína se dirige hacia el núcleo y reconoce el sitio correcto de unión en la envoltura nuclear. En un segundo paso, el grupo de los 3 aminoácidos terminales (-AAX) se separa de la prelámina quedando como aminoácido terminal la cisteína farnesilada. Este paso se realiza en la membrana del retículo endoplasmático por acción de la metaloproteinasa Zmpste24, con sus sitios activos hacia el citoplasma. El tercer paso consiste en la metilación de la cisteína farnesilada por la acción de la isoprenilcisteína carboxil metiltransferasa también localizada en el retículo endoplasmático. Posteriormente, a través de los poros nucleares, las importinas translocan la prelámina-A al núcleo, donde finalmente experimenta un corte proteolítico por acción nuevamente de la enzima Zmpste24. La prelámina-A se corta en dos segmentos: uno que contiene los últimos 15 aminoácidos de la prelámina-A y que es degradado (incluida la cisteína farnesilada y metilada) y el otro, forma la lámina-A madura que se fija a la envoltura nuclear interna por medio de diversas proteínas de unión (Barrowman, et al., 2008).
En la progeria infantil, la lámina-A alterada más corta sufre los cambios de farsenilación, metilación y su translocación al núcleo, pero su segmento terminal de 15 aminoácidos no puede cortarse por la enzima Zmpste24. Esto es debido a que las secuencias para realizar el corte y liberación de la proteína madura están ausentes y por lo tanto permanece farnesilada y origina una proteína anormal que se denomina progerina (figura 2) (De Sandre-Giovannoli et al., 2003). La progerina se acumula en el núcleo y altera la estructura de la envoltura nuclear, y consiguientemente la función nuclear y del fenotipo de este raro síndrome (Cao, et al., 2007; González y Andrés, 2011; Reddy y Comai, 2012).
. A la derecha se muestra la síntesis de la progerina, cuya acumulación es la causante del HGPS. (modificado de Musich y Zou, 2009).")
Vía de síntesis de la lamina-A (izquierda). A la derecha se muestra la síntesis de la progerina, cuya acumulación es la causante del HGPS. (modificado de Musich y Zou, 2009).
En 1998, los doctores Leslie Gordon y Scott Berns crearon la fundación para la investigación de la progeria (PRF, de sus siglas en inglés) tras enfrentarse al diagnóstico de HGPS de su hijo Sam de 22 meses de edad. La falta de información de la enfermedad, la ausencia de tratamiento y de investigación científica orientada a descubrir las causas del síndrome y una potencial cura, motivaron a los doctores y a un grupo de colegas para fundar la PRF. Esta organización, única en el mundo, realiza investigación acerca de la progeria, promueve el conocimiento de la enfermedad y recauda fondos para los proyectos de investigación. La PRF cuenta con un banco de células y tejidos de pacientes con progeria y de sus familiares para las investigaciones de la progeria y otras enfermedades relacionadas con el envejecimiento. Actualmente, la PRF tiene un registro de 49 líneas celulares de ninos con progeria alrededor de todo el mundo. Las edades de estos niños varían entre 8 meses y 17 años de edad. También se tiene el registro de 36 líneas de células de sus familiares más inmediatos.
Los miembros del Consorcio Genético y las líneas celulares del banco de células de la PRF fueron esenciales para los experimentos que condujeron al descubrimiento del gen causante de la progeria, un paso necesario para la búsqueda de la cura. El número de científicos interesados en investigar la progeria ha aumentado gracias al descubrimiento del gen. Este primer objetivo culminó en 2003 con la identificación de la mutación del gen LMNA como causante de la enfermedad (De Sandre-Giovannoli, et al., 2003; Eriksson, et al., 2003). El descubrimiento del gen causante del HGPS y el conocimiento de la patogenia molecular de la enfermedad ha propiciado investigaciones en modelos animales para profundizar en el conocimiento de la progeria.
Experimentación animalMás allá de los estudios con cultivos celulares, los investigadores consiguieron reproducir la enfermedad usando modelos murinos modificados genéticamente en los que se sustituyó la secuencia del gen LMNA normal codificante de la lámina-A por una idéntica a la que portan los enfermos con HGPS; es decir con la mutación que impide el correcto procesamiento de la lámina-A. Al igual que ocurre en la HGPS, los ratones así modificados nacen sanos, pero inmediatamente comienzan a manifestar los mismos síntomas que se observan en los humanos (Mounkes, et al., 2003).
Otro modelo animal, se basó en la importancia de la enzima Zmpste24 para el correcto procesamiento de la lámina-A. Se emplearon ratones deficientes en la proteasa Zmpste24 (ratones knockout Zmpste24−/−) para analizar los efectos de su ausencia. En los animales experimentales esta ausencia determina la acumulación de una pre-lámina A farnesilada que no es cortada en ninguno de los segmentos de aminoácidos. Los resultados demostraron que estos animales tienen apariencia normal hasta las tres semanas de vida posnatal, momento en el cual comienzan a manifestar un proceso de envejecimiento prematuro, idéntico a los niños con HGPS, caracterizado por alopecia, cifosis, osteólisis, deficiencias dentales, debilidad muscular y artritis, y suelen morir entre las 24 y 32 semanas. El análisis de la proteína lámina-A reveló el mismo tipo de acumulación de progerina, la forma no procesada de lámina-A, el mismo defecto que se observa en los niños con HGPS. Esto llevo a identificar la proteasa, la “tijera molecular” responsable del procesamiento normal de la pre-lámina A, que no es otra que la Zmpste24 (Varela, et al., 2005).
Para comprobar si la pre-lámina A farnesilada era el verdadero agente causante de las alteraciones nucleares se desarrolló un ratón tipo mixto (ratón Zmpste24−/− Lmna+/−), es decir, no expresa la proteína Zmpste24, pero por otra parte es heterocigoto para LMNA y expresa el 50% de la lámina-A farnesilada. La comparación de este ratón con el knockout exclusivo (Zmpste24−/−) mostró que los fibroblastos de los ratones mixtos (ratón Zmpste24−/− Lmna+/−) tenían menos alteraciones en la morfología del núcleo celular y disminución en la cantidad de prelámina-A. Esto sugiere que la acumulación de la pre-lámina A farnesilada puede ser la causa de las alteraciones fenotípicas en los núcleos celulares asociados al HGPS, y que cuando se disminuye la expresión del gen LMNA, se reducen los efectos tóxicos de esta proteína (Fong, et al., 2004).
Basándose en estos resultados no solo se conocieron más detalles del proceso normal de la pre-lámina A, si no que se dispone de un nuevo modelo animal en el que se puede estudiar el desarrollo de la enfermedad, y así poder realizar pruebas terapéuticas que permitan en primer lugar, revertir las alteraciones morfológicas de las células in vitro de estos animales; y en un segundo lugar, actuar sobre el animal completo, para finalmente usar estos resultados en humanos y poder utilizar terapias para su tratamiento.
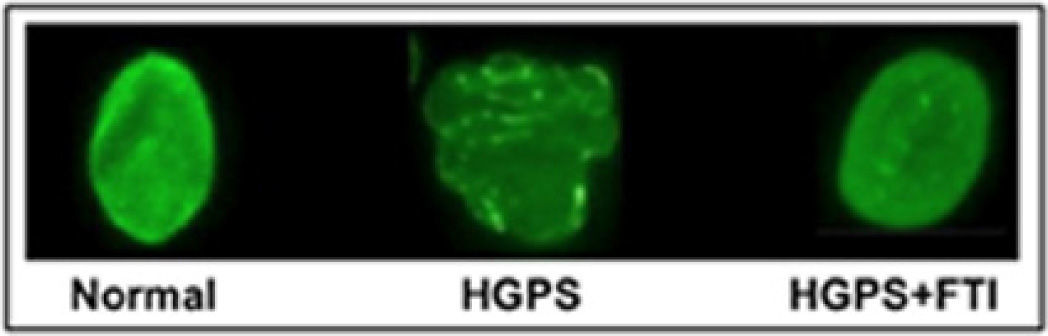
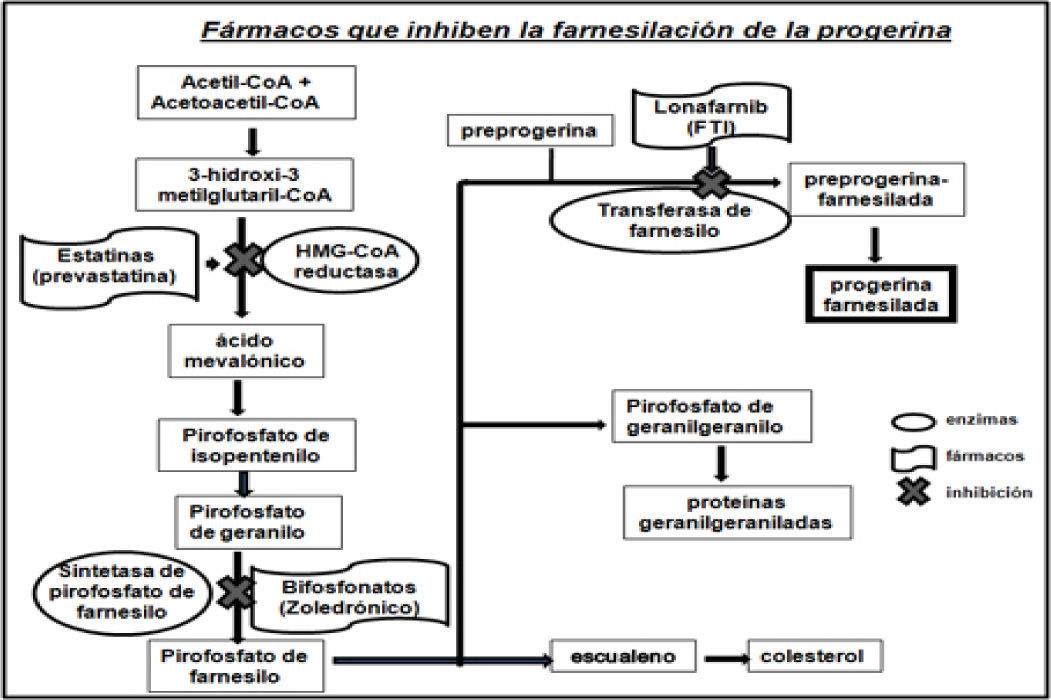
Fármacos que inhiben la farnesilación de la progerinaEl fármaco lonafarnib es un inhibidor de las farnesil-transferasa (FTI), y no permite que se una el grupo farnesilo a la progerina. Varios investigadores probaron el efecto del lonafarnib en cultivos celulares y en modelos animales con HGPS, logrando revertir las alteraciones de la estructura nuclear (figura 3), así como mejorar la condición cardiaca de los ratones tratados con el FTI al compararlos con ratones no tratados. El fármaco impide que la proteína anormal (progerina) se una al núcleo de las células y así los síntomas de la progeria disminuyen. Otras investigaciones pusieron de manifiesto la posibilidad de mejorar este tratamiento experimental a base de lonafarnib mediante la combinación con prevastatina y ácido zoledrónico (figura 4).
, de HGPS (centro) y de HGPS + FTI (derecha) (tomada de Capell, et al., 2005).")
Aspecto del núcleo celular normal (Izquierda), de HGPS (centro) y de HGPS + FTI (derecha) (tomada de Capell, et al., 2005).
.")
Fármacos que inhiben la farnesilación de la progerina (modificado de Baek, et al., 2013).
La prevastatina pertenece a la clase de fármacos denominados estatinas, comúnmente se usa para reducir los niveles de colesterol y prevenir enfermedades cardiovasculares. Por lo general, los niños con progeria no tienen colesterol alto, pero este fármaco se utiliza porque inhibe la enzima 3-hidroxi-3-metilglutaril-coenzima A (HMG-CoA) reductasa bloqueando la síntesis de la molécula de farnesilo, y el ácido zoledrónico es un bifosfonato, utilizado para la osteoporosis (Fong, 2006; Varela, et al., 2008), debido a que los niños con progeria tienen baja densidad ósea este fármaco puede ayudar a mejorarla con el tiempo. El ácido zoledrónico inhibe la enzima farnesil-pirofosfato-sintetasa y al igual que la prevastatina bloquea la síntesis de la molécula de farnesilo (figura 4). La combinación de estos tres fármacos en modelos de ratones demostró su eficacia al mejorar el aspecto de los animales e incrementar su esperanza de vida.
Verstraeten y colaboradores demostraron efectos negativos al tratamiento con FTI, el cual induce la formación de núcleos en forma de dona tanto in vitro en células no transformadas y tumorales como in vivo en células intestinales y de piel de ratones tratados con FTI por 6–12 meses. Estas células con núcleos en forma de dona presentan defectos en la cariocinesis, desarrollan aneuploidias (cambio en el número cromosómico) y a menudo son binucleadas con proliferación lenta, lo que se ha atribuido a un defecto en la separación del centrosoma y a los niveles bajos de pericentrina (proteína del centrosoma, esencial para la correcta división celular), ya que el tratamiento con FTI incrementa la degradación proteosomal de esta proteína (Verstraeten et al., 2011) e inhibe la farnesilación de la lámina B1 y B2, provocando consecuencias en las funciones celulares (Adam, et al., 2013; Wang, 2012) y defectos en el desarrollo del cerebro y notables anormalidades nucleares en neuronas (Jung, et al., 2013). Cuando se utilizan la combinación de 3 FTIs distintos inducen más núcleos en forma de dona que cuando se utiliza un solo FTI (Verstraeten, et al., 2011).
Aunque se ha reportado que los ninos con HGPS que tomaron el FTI dos veces al día presentaron efectos secundarios leves como diarrea y cambios en algunas pruebas de función hepática, deben ser revisados cuidadosamente, e ir modificando la dosis del fármaco según aparezcan efectos secundarios inesperados.
Posibles vías terapéuticas en el HGPSUna de ellas es la terapia antisentido que consiste en la utilización de un oligonucleótido diseñado para ser parte complementaria (antisentido) de determinado ARNm, para evitar la expresión de la correspondiente proteína y con ella la actividad del gen involucrado. En el caso del HGPS se utiliza un oligonucleótido sinsentido que contiene la secuencia complementaria del exón 11, para evitar el corte del ARNm mutado que produce la progerina y así evitar la producción de esta proteína mutada. Se ha demostrado que estos oligonucleótidos sinsentido cuando se añaden a fibroblastos de pacientes con HGPS, reducen la expresión del ARNm mutado y, en consecuencia, disminuye la producción de progerina y las alteraciones morfológicas del núcleo celular (Scaffidi y Misteli, 2005). Usando modelos animales con terapia de oligonucleótidos sinsentido, se comprobó que los ratones mutantes, disminuyen la acumulación de progerina, mejoran el fenotipo progeroide y aumentan su sobrevida (Osorio, et al., 2011). Estos resultados son el primer paso hacia el desarrollo de un futuro ensayo clínico con oligonucleótidos antisentido en pacientes con HGPS.
También recientemente la rapamicina ha ganado su atención como nuevo candidato para el tratamiento del HGPS. La rapamicina (también conocido como sirolimus), es un inmunosupresor que se utiliza para evitar el riesgo de rechazo de órganos en pacientes que recibieron trasplantes. Recientes investigaciones demostraron que la rapamicina puede prolongar la vida en ratones sanos (Harrison, et al., 2009; Miller, et al., 2010), y reducir lesiones ateroescleróticas en ratones a pesar de severa hipercolesterolemia, incluso cuando los ratones tienen una dieta rica en grasas (Waksman, et al., 2003). Además se ha reportado que la rapamicina elimina la progerina a través de la autofagia y revierte el fenotipo celular en fibroblastos de niños con HGPS, y en consecuencia retrasa el envejecimiento celular (Cao, et al., 2011A); Graziotto, et al., 2012). Estos resultados siguieren los beneficios de la rapamicina en la longevidad, pero sus efectos deben ser estudiados cuidadosamente en modelos de ratón con HGPS, para ser considerada como una posible terapia para pacientes con HGPS. Ya que se ha reportado que el tratamiento con rapamicina en pacientes trasplantados provoca efectos secundarios como: sintomas gastrointestinales, edema, infección, niveles altos de colesterol y triglicéridos, anemia y neumonitis intersticial (Baur, et al., 2011). Otro inconveniente para usar la rapamicina en ninos con HGPS es el efecto sobre el crecimiento y desarrollo ya que este fármaco inhibe la proteína mTOR (siglas en inglés de mammalian target of rapamycin), el cual es un regulador del crecimiento y proliferación celular.
Sin embargo, considerando la severidad de la enfermedad, los beneficios pueden compensar los efectos secundarios, y quizás se pueda controlar con dosis y horarios muy controlados, por ejemplo periodos intermitentes de tratamiento, seguidos por periodos de recuperación, más que tratamientos crónicos. Se ha considerado la co-administración de rapamicina con FTIs en tratamientos clínicos para el HGPS, ya que la rapamicina elimina la progerina farnesilada por autofagia, y los FTIs limitan la producción de progerina farnesilada en primer lugar.
Pruebas clínicas en humanosCon base en los resultados obtenidos en estudios celulares y ratones HGPS con fármacos que inhiben la farnesilación de la progerina, en mayo del 2007 se inicio un período de pruebas clínicas en niños con progeria utilizando lonafarnib como (FTI). El ensayo finalizó en diciembre del 2009 y los resultados se publicaron en octubre del 2012 (Gordon, et al., 2012). En este ensayo participaron 26 niños de 16 países, los niños recibieron dos veces al día lonafarnib por vía oral durante dos anos y medio. Los ni'os que terminaron su tratamiento mostraron mejoría en uno o varios aspectos como el aumento de peso, la rigidez esquelética y sobre todo en su sistema cardiovascular. Con estos resultados se pudo demostrar que el lonafarnib no solo puede retrasar el daño en los vasos sanguíneos, sino también revertirse parcialmente, en tan sólo 2.5 anos de tratamiento, lo que representa un gran avance porque las enfermedades cardiovasculares son la principal causa de muerte en los niños con progeria. Posteriormente se han iniciado dos estudios clínicos adicionales de tratamiento en ninos con progeria. En Francia, desde el 2008 se están tratando niños con HGPS con los fármacos prevastatina y ácido zoledrónico. Otro estudio en Boston comenzó en el 2009, y en él están tratando a los niños con los tres fármacos (lonafarnib, prevastatina y ácido zoledrónico). La fundación para la investigación de la progeria espera que las tres drogas actúen en conjunto, complementándose y puedan proporcionar la oportunidad de ampliar la eficacia sobre los fármacos usados individualmente. En la primera etapa se aplicó a cinco niños y como sus efectos secundarios fueron tolerados, ahora el equipo se aboca a ampliar el ensayo a 45 pacientes de 19 países (Young, et al., 2013).
Envejecimiento y progerinaAlgunas investigaciones han demostrado que la proteína progerina también se produce en las células normales, y esta producción aumenta en grandes niveles, a medida que se acerca la senectud (Scaffidi y Misteli, 2006; Rodriguez, et al., 2009; Burtner y Kennedy, 2010). Varios estudios han vinculado la progerina con el envejecimiento normal, incluyendo el vínculo entre la progerina y la inestabilidad genética, principalmente la disfunción de los telómeros (fragmentos de ADN que se encuentran en los extremos de los cromosomas) en el proceso de envejecimiento. Los telómeros se desgastan durante la división celular, cuando el grado de desgaste es elevado la célula deja de dividirse y muere. Estos telómeros cortos y disfuncionales son los que activan la producción de progerina, lo que se asocia con el daño celular relacionado con la edad. Al tiempo que los telómeros se acortan como consecuencia de la división celular, se produce más progerina (Cao, et al., 2011; González-Morán, 2012). Seguir investigando sobre el efecto de los fármacos que inhiben la farnesilación de la progerina no sólo abrirá una esperanza en el tratamiento de la progeria infantil, sino que puede proporcionar claves para tratar a millones de adultos con enfermedades cardiacas y accidentes cerebro vasculares asociados con el proceso normal de envejecimiento (Olive, et al., 2010). Es de esperar que los tratamientos que se apliquen para la progeria sirvan también en parte para prolongar los años de vida útil del adulto, deteniendo el envejecimiento fisiológico, especialmente el vascular.
Sin duda la progeria infantil es una enfermedad realmente cruel, son niños que padecen y sufren los mismos padecimientos que los ancianos, pero el desarrollo de la inteligencia no se ve afectado. No debemos olvidar que hay un niño detrás de las arrugas, la calvicie, atrapado en un cuerpo fatigado, que no sobrepasa el metro de estatura y que le agobian los dolores, pero que le gusta jugar y reír, como cualquier niño, porque es un niño en el cuerpo de un anciano, y saben encontrar el sentido de la vida. Pensar que un pequeño error ortográfico genético, en una sola letra entre 3 mil millones de letras de la herencia humana, sea el responsable de un efecto tan terrible en el cuerpo. Estos niños nos obligan a reflexionar sobre lo afortunados que somos todos aquellos que disponemos de buena salud.