




Las actuales guías de práctica clínica recomiendan considerar el estudio genético en todos los pacientes con diagnóstico de feocromocitoma o paraganglioma (PPGL). El objetivo de nuestro trabajo es conocer el porcentaje de solicitud de estudio genético en el PPGL en un hospital universitario no especializado, los factores implicados en dicha solicitud, cuántos de ellos presentan mutación germinal, cuáles son los genes afectados y qué variables se relacionan con la presencia de mutaciones.
Material y métodosSe incluyó a todos los pacientes con PPGL diagnosticados en el área sanitaria de un hospital universitario de tercer nivel entre 2010 y 2015. Se recogieron las variables: edad, localización, único o múltiple, secreción hormonal, cuadro sindrómico, antecedentes familiares y servicio responsable. Se comparó a los pacientes con estudio genético realizado (GEN+) frente a aquellos no estudiados (GEN–), y pacientes con mutación (MUT+) frente a aquellos sin mutación predisponente (MUT–).
ResultadosSe incluyó a 39 pacientes (21 mujeres y 18 varones con edad media 53,9±17,8 años). Se hizo estudio genético al 54% y estos eran más jóvenes, con antecedentes familiares, múltiples, secretores y con mayor frecuencia vistos por Endocrinología. Hubo menos paragangliomas de cabeza y cuello unilaterales. Un 33% tenía mutación germinal (3 RET, 3 SDHB, un SDHD) y estos eran más jóvenes, más sindrómicos, múltiples o con antecedentes familiares.
ConclusionesAunque las guías de práctica clínica recomiendan considerar la realización de un estudio genético a todos los pacientes con PPGL, en nuestra Área de Salud se solicitó en el 54% de ellos. Un 33% de ellos presentaron mutación germinal predisponente.
Current clinical practice guidelines recommend that a genetic study is considered in all patients diagnosed with pheochromocytoma or paraganglioma (PPGL). Our study objective was to know how many patients with PPGL undergo genetic studies at a non-specialized university hospital, the clinical factors involved in the decision to make the study, how many patients are found germline mutation, which are the affected genes, and what variables are related to presence of mutations.
Material and methodsAll patients diagnosed with PPGL at a tertiary university hospital from 2010 to 2015 were enrolled. Age and sex, tumor location and multiplicity, hormone secretion, presence of a clinical syndrome, family history, and medical department in charge were recorded and used to compare patients with (GEN+) and without (GEN-) genetic study, as well as patients with (MUT+) and without (MUT-) germline mutations.
ResultsThirty-nine patients were enrolled (21 females and 18 males with a mean age of 53.9±17.8 years). A genetic study was performed in 54% of patients with PPGL. These were younger, were more frequently seen by endocrinologists, and had more often a family history related to PPGL, multiple PPGLs, or hormonally functional tumors. Unilateral head and neck paragangliomas were less common. Germline mutations (3 RET, 3 SDHB, 1 SDHD) were found in 33% of patients, who were younger and more frequently had a clinical syndrome, multiple PPGLs. and a family history of PPGL.
ConclusionAlthough current clinical practice guidelines recommend that genetic studies are considered in all patients diagnosed with PPGL, studies was requested for 54% of such cases in our healthcare area. Predisposing germline mutations were found in 33% of studies.
El feocromocitoma y el paraganglioma (PPGL) son tumores derivados del tejido cromafín. La mayoría de estos tumores producen catecolaminas responsables de la sintomatología simpaticomimética, siendo la hipertensión arterial el hallazgo más habitual. Los paragangliomas de cabeza y cuello, de estirpe parasimpática, no suelen secretar catecolaminas, por lo que no producen hipertensión y se diagnostican habitualmente por efecto masa, como incidentalomas, o durante el seguimiento de los portadores de alguna mutación predisponente.
Desde 1990, se han descubierto al menos 15 genes de susceptibilidad para PPGL. Las mutaciones en 3 de ellos se asocian a cuadros sindrómicos: von Hipple-Lindau (VHL), MEN2 (RET) y neurofibromatosis tipo 1 (NF1). Otros 5 genes participan en la conformación de la enzima succinato deshidrogenasa de la mitocondria: 2 genes codifican sus subunidades catalíticas (SDHA y SDHB), otros 2 las subunidades de anclaje (SDHD y SDHC), mientras que el gen SDHAF2 codifica la proteína responsable de la flavinización de SDHA, necesaria para un correcto ensamblaje de la enzima. Sus mutaciones se implican sobre todo en el desarrollo de los paragangliomas. Otros genes de susceptibilidad descritos en pocos casos son TMEM127, MAX, FH, EGLN1/PHD2, KIF1β, IDH1 y HIF2α/EPAS11-3. Los PPGL pueden agruparse en 2 clústeres en base a su perfil de expresión génica. El clúster 1 agrupa aquellos tumores con mutaciones en los genes que participan en la respuesta a la hipoxia (SDHx [cluster 1A] y VHL [cluster 1B]): sus mutaciones activan estas vías y la angiogénesis en situación de normoxia. El clúster 2 incluye los tumores con mutaciones en los genes RET, NF1, TMEM127 y MAX, todos ellos implicados en la activación de las cascadas de señalización intracelular MAPK y PI3K-AKT-mTOR y en la replicación celular2-4.
Las guías actuales1 recomiendan que todos los pacientes reciban información acerca del diagnóstico genético y participen en la toma de decisiones sobre la conveniencia de su realización, pero no se han realizado estudios que evalúen el grado de cumplimiento de esta recomendación en la práctica clínica real en distintos ámbitos. El abordaje propuesto en dichas guías es el análisis secuencial de genes comenzando por los más probablemente mutados teniendo en cuenta las características clínicas de cada caso y la frecuencia relativa de los distintos genes, hasta encontrar una mutación patogénica. Sin embargo, este abordaje resulta costoso en términos de tiempo y de dinero, y varios grupos han comunicado excelentes resultados utilizando paneles next generation sequencing (NGS), que estudian simultáneamente la práctica totalidad de los genes conocidos predisponentes a PPGL con un menor coste5-7.
Varios grupos de trabajo dedicados a la genética de feocromocitomas y paragangliomas, que han publicado en años recientes el porcentaje de pacientes con PPGL portadores de alguna mutación germinal predisponente, han comunicado resultados entre el 14 y el 41%5,6,8-14. Sin embargo, la mayoría de estas publicaciones vienen de centros de referencia para el estudio genético, que reciben muestras de múltiples hospitales, por lo que no pueden asegurar estar estudiando a la totalidad de los pacientes que son diagnosticados de PPGL. Es posible que el estudio genético no se haya realizado en algunos pacientes con baja probabilidad de mutación germinal (tumores suprarrenales, únicos, no sindrómicos, en pacientes de mayor edad y sin antecedentes familiares), siendo más probable su solicitud en pacientes de mayor riesgo, incrementando de forma espuria el porcentaje de pacientes con mutaciones.
El objetivo del presente trabajo es conocer a qué porcentaje de pacientes diagnosticados de PPGL y atendidos en un hospital universitario no especializado se les realiza un estudio genético, qué factores se relacionan con la solicitud de dicho estudio, cuántos de ellos son portadores de una mutación germinal, cuáles son los genes afectados y qué variables se relacionan con la presencia de mutaciones. Como objetivo secundario, se pretende determinar cuántos pacientes con PPGL son seguidos en otras especialidades diferentes de Endocrinología y, en tal caso, si existe uniformidad en los criterios de solicitud de estudio genético en estos pacientes con respecto a los que son atendidos por Endocrinología.
Material y métodosSe revisaron las historias clínicas de todos los pacientes con un diagnóstico de PPGL en los informes de alta o en los informes de anatomía patológica entre el 1 de enero del 2010 y el 31 de diciembre del 2015 en un hospital universitario de tercer nivel. Se incluyó a todos aquellos con confirmación anatomopatológica del diagnóstico (38 pacientes) y una paciente no intervenida, pero con un claro diagnóstico clínico, bioquímico y radiológico. Tres pacientes (todos ellos con paragangliomas de cabeza y cuello) fueron intervenidos en otro hospital. La obtención de datos a partir de la codificación tanto de informes de alta como de informes de anatomía patológica permite asegurar la recuperación de la práctica totalidad de los pacientes con esta patología.
Las variables analizadas fueron: edad al diagnóstico, sexo, localización del tumor, multiplicidad, secreción hormonal (metanefrinas totales y fraccionadas en orina de 24 h mediante HPLC), cuadro sindrómico asociado, antecedentes familiares de PPGL, servicio médico a cargo del paciente, realización o no de estudio genético y mutaciones germinales encontradas. Estas variables se utilizaron para comparar a los pacientes con estudio genético realizado (GEN+) frente a aquellos no estudiados (GEN–), así como, dentro de los pacientes con estudio genético, a los pacientes con mutación predisponente a PPGL (MUT+) frente a aquellos en los que no se encontró mutación alguna (MUT–). La variable «sexo» solo se utilizó para el análisis descriptivo de la población y la variable «secreción hormonal» no se tuvo en cuenta cuando se compararon MUT+frente a MUT–.
El estudio genético se realizó en muestras de sangre periférica. La mayoría de las muestras fueron remitidas al Grupo de Cáncer Endocrino Hereditario del Centro Nacional de Investigaciones Oncológicas (CNIO) de Madrid. Hasta 2014, se estudiaron mediante técnicas de Sanger y multiplex ligand-dependent probe amplification (MLPA)10,14-16 los distintos genes de manera secuencial, comenzando por los más probablemente mutados teniendo en cuenta las características clínicas de cada caso y la frecuencia relativa de los distintos genes, hasta encontrar una mutación patogénica. Los casos más recientes fueron estudiados mediante un panel NGS7.
El análisis estadístico se realizó con el programa G-Stat 2.0. Para la variable cuantitativa edad utilizamos la media ± desviación típica como parámetros descriptivos, y para realizar la inferencia poblacional entre los grupos el test no paramétrico U de Mann-Whitney al presentar todos los subgrupos n <30. Para el resto de las variables, todas ellas cualitativas, se usaron proporciones y los subgrupos se compararon con el test de la chi al cuadrado o test exacto de Fisher (este último si alguna de las casillas tenía menos de 5 sujetos).
El presente estudio ha recibido la aprobación de la Comisión de Investigación y el Comité Ético de Investigación Médica de nuestro hospital.
ResultadosAnálisis descriptivo en nuestra poblaciónDel total de 39 pacientes, 21 eran mujeres (53,8%) y 18 varones (46,5%). La media de edad al diagnóstico fue de 53,9±17,8 años.
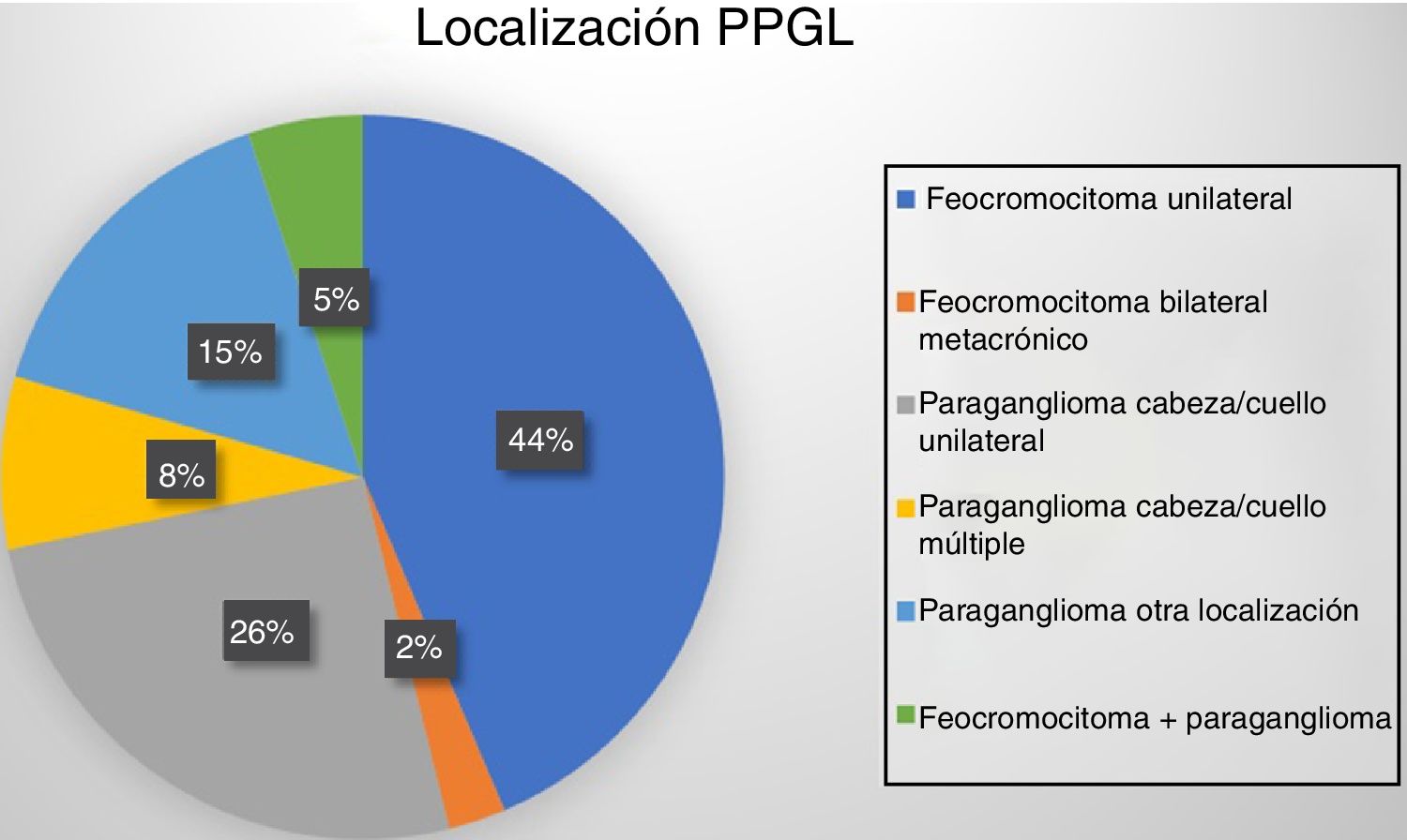
Dieciocho pacientes (47%) presentaron tumoración suprarrenal, uno de ellos bilateral metacrónico (un paciente con síndrome MEN2A). Otros 19 casos (48%) presentaron paragangliomas: 13 en cabeza y cuello (10 únicos, uno recurrente y 2 bilaterales) y los 6 restantes fueron toracoabdominales (uno de ellos recurrente y otro maligno). Por último, 2 pacientes desarrollaron paragangliomas tras la resección años antes de un feocromocitoma (fig. 1).

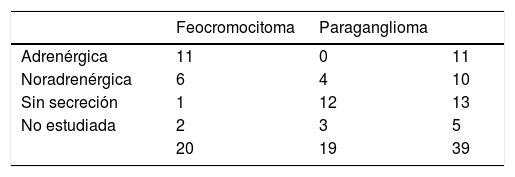
Diecisiete de los 20 feocromocitomas (85%) eran secretores, 11 adrenérgicos y 6 noradrenérgicos. De los 3 restantes, uno lo consideramos no secretor, al tratarse de un varón de 32 años en seguimiento por carcinoma medular de tiroides y portador de mutación en RET, en el que apareció un incidentaloma suprarrenal de un centímetro indicativo de feocromocitoma, con metanefrinas urinarias normales. El estudio histológico del mismo fue compatible con feocromocitoma. En los otros 2 no disponíamos del dato, uno de ellos por haber sido estudiado en otro hospital. De los 19 paragangliomas, 4 (21%) presentaban un patrón de secreción noradrenérgico, 12 eran no secretores y en 3 no se había realizado la determinación prequirúrgica de metanefrinas por no sospecharse PPGL en primera instancia (tabla 1).
Dos pacientes referían antecedentes familiares de paragangliomas y 3 pacientes de carcinoma medular de tiroides. El resto no presentaba antecedentes familiares de interés, o al menos no habían sido recogidos en la historia clínica.
Solo uno de los 39 casos del estudio presentó un comportamiento maligno. Era una mujer diagnosticada a los 23 años de un paraganglioma interaorto-cava que posteriormente desarrolló metástasis hepáticas y óseas. Los estudios genéticos realizados hasta la fecha no han encontrado mutaciones germinales y sí una mutación somática en el gen NF1.
En nuestra serie, 23 pacientes (59%) fueron atendidos en Endocrinología (15/18 feocromocitomas, 2/2 paraganglioma+feocromocitoma, 4/6 paragangliomas toraco-abomino-pélvicos y 2/13 paragangliomas de cabeza/cuello), 9 casos en Cirugía Vascular (23%), todos ellos paragangliomas únicos de cabeza y cuello no secretores, y los 7 restantes (18%) en Otorrinolaringología, Nefrología, Medicina Interna, Urología y Neurocirugía.
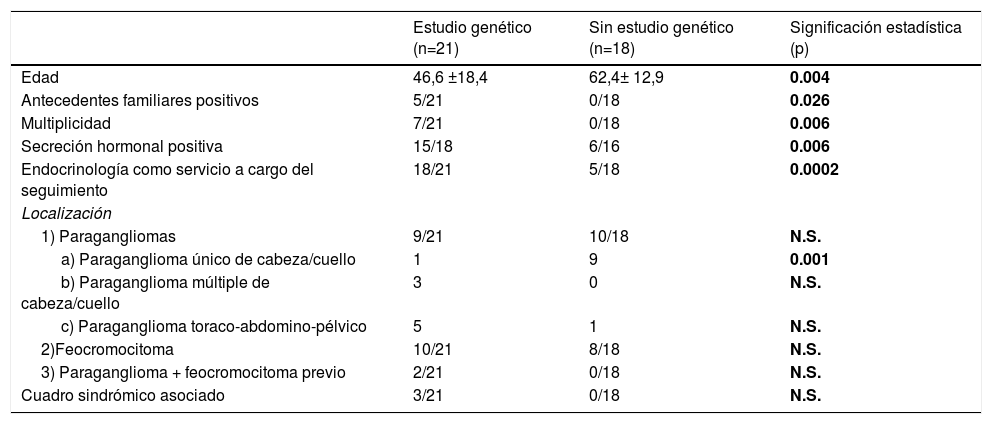
¿Quiénes fueron los pacientes con PPGL sometidos a estudio genético y qué les hace diferentes con respecto a los no estudiados?El estudio de mutaciones germinales se realizó en 21 pacientes (GEN+54%), que se compararon con los pacientes no estudiados (GEN–). Los pacientes estudiados eran significativamente más jóvenes, tenían más antecedentes familiares, con mayor frecuencia eran múltiples (bilaterales o recurrentes), secretores y habían sido atendidos por Endocrinología, y con menor frecuencia eran tumores únicos de cabeza y cuello (tabla 2). El estudio genético se realizó en todos los pacientes sindrómicos, aunque la diferencia con los pacientes no estudiados no alcanzó significación estadística, probablemente debido a nuestro pequeño tamaño muestral.
Comparativa entre los pacientes de la serie estudiados genéticamente (n=21) frente a los casos de PPGL sin estudio genético (n=18)
| Estudio genético (n=21) | Sin estudio genético (n=18) | Significación estadística (p) | |
|---|---|---|---|
| Edad | 46,6 ±18,4 | 62,4± 12,9 | 0.004 |
| Antecedentes familiares positivos | 5/21 | 0/18 | 0.026 |
| Multiplicidad | 7/21 | 0/18 | 0.006 |
| Secreción hormonal positiva | 15/18 | 6/16 | 0.006 |
| Endocrinología como servicio a cargo del seguimiento | 18/21 | 5/18 | 0.0002 |
| Localización | |||
| 1) Paragangliomas | 9/21 | 10/18 | N.S. |
| a) Paraganglioma único de cabeza/cuello | 1 | 9 | 0.001 |
| b) Paraganglioma múltiple de cabeza/cuello | 3 | 0 | N.S. |
| c) Paraganglioma toraco-abdomino-pélvico | 5 | 1 | N.S. |
| 2)Feocromocitoma | 10/21 | 8/18 | N.S. |
| 3) Paraganglioma + feocromocitoma previo | 2/21 | 0/18 | N.S. |
| Cuadro sindrómico asociado | 3/21 | 0/18 | N.S. |
NS: no significativa.
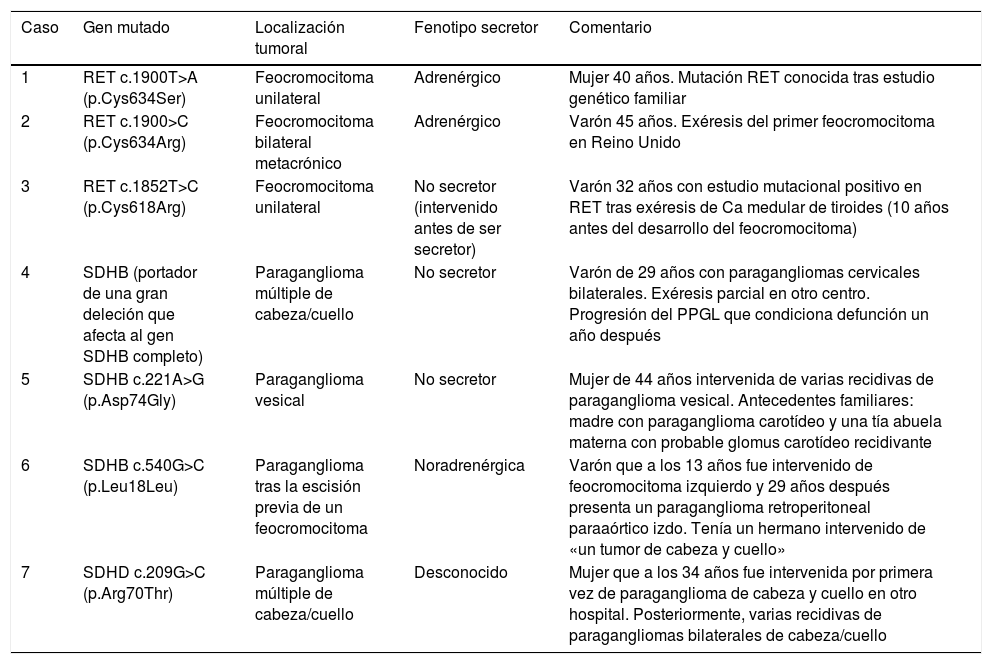
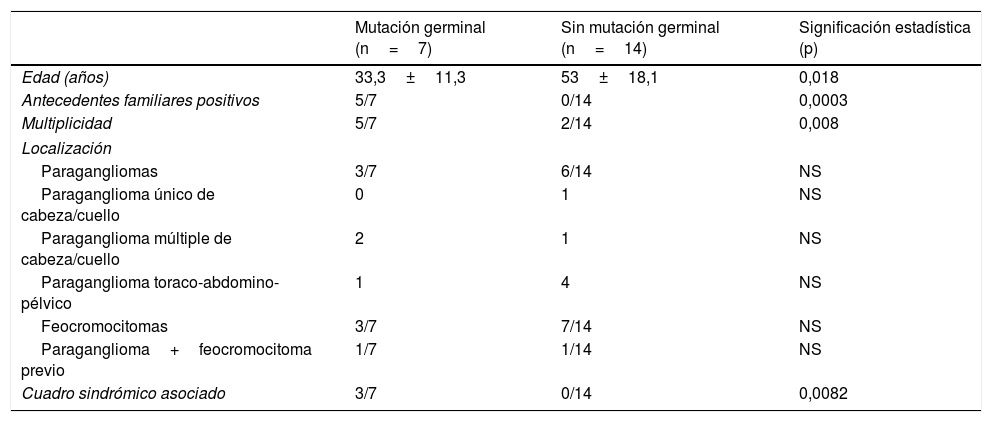
De los 21 pacientes con estudio genético realizado, 7 (33%) presentaban mutaciones germinales: 3 en RET, 3 en SDHB y uno en SDHD. Sus características clínicas se describen en la tabla 3.
Características clínicas de los pacientes portadores de mutaciones germinales predisponentes a PPGL
| Caso | Gen mutado | Localización tumoral | Fenotipo secretor | Comentario |
|---|---|---|---|---|
| 1 | RET c.1900T>A (p.Cys634Ser) | Feocromocitoma unilateral | Adrenérgico | Mujer 40 años. Mutación RET conocida tras estudio genético familiar |
| 2 | RET c.1900>C (p.Cys634Arg) | Feocromocitoma bilateral metacrónico | Adrenérgico | Varón 45 años. Exéresis del primer feocromocitoma en Reino Unido |
| 3 | RET c.1852T>C (p.Cys618Arg) | Feocromocitoma unilateral | No secretor (intervenido antes de ser secretor) | Varón 32 años con estudio mutacional positivo en RET tras exéresis de Ca medular de tiroides (10 años antes del desarrollo del feocromocitoma) |
| 4 | SDHB (portador de una gran deleción que afecta al gen SDHB completo) | Paraganglioma múltiple de cabeza/cuello | No secretor | Varón de 29 años con paragangliomas cervicales bilaterales. Exéresis parcial en otro centro. Progresión del PPGL que condiciona defunción un año después |
| 5 | SDHB c.221A>G (p.Asp74Gly) | Paraganglioma vesical | No secretor | Mujer de 44 años intervenida de varias recidivas de paraganglioma vesical. Antecedentes familiares: madre con paraganglioma carotídeo y una tía abuela materna con probable glomus carotídeo recidivante |
| 6 | SDHB c.540G>C (p.Leu18Leu) | Paraganglioma tras la escisión previa de un feocromocitoma | Noradrenérgica | Varón que a los 13 años fue intervenido de feocromocitoma izquierdo y 29 años después presenta un paraganglioma retroperitoneal paraaórtico izdo. Tenía un hermano intervenido de «un tumor de cabeza y cuello» |
| 7 | SDHD c.209G>C (p.Arg70Thr) | Paraganglioma múltiple de cabeza/cuello | Desconocido | Mujer que a los 34 años fue intervenida por primera vez de paraganglioma de cabeza y cuello en otro hospital. Posteriormente, varias recidivas de paragangliomas bilaterales de cabeza/cuello |
Estos 7 pacientes eran significativamente más jóvenes, presentaban con mayor frecuencia cuadros sindrómicos y tenían más antecedentes familiares y más tumores múltiples. No hubo diferencias significativas en la localización de los tumores entre pacientes con y sin mutación germinal. Estos datos se presentan en la tabla 4.
Comparativa entre los pacientes con estudio mutacional germinal positivo (n=7) frente a aquellos sin mutación en la línea germinal (n=14)
| Mutación germinal (n=7) | Sin mutación germinal (n=14) | Significación estadística (p) | |
|---|---|---|---|
| Edad (años) | 33,3±11,3 | 53±18,1 | 0,018 |
| Antecedentes familiares positivos | 5/7 | 0/14 | 0,0003 |
| Multiplicidad | 5/7 | 2/14 | 0,008 |
| Localización | |||
| Paragangliomas | 3/7 | 6/14 | NS |
| Paraganglioma único de cabeza/cuello | 0 | 1 | NS |
| Paraganglioma múltiple de cabeza/cuello | 2 | 1 | NS |
| Paraganglioma toraco-abdomino-pélvico | 1 | 4 | NS |
| Feocromocitomas | 3/7 | 7/14 | NS |
| Paraganglioma+feocromocitoma previo | 1/7 | 1/14 | NS |
| Cuadro sindrómico asociado | 3/7 | 0/14 | 0,0082 |
NS: no significativa.
Aunque las guías actuales de práctica clínica recomiendan plantear el estudio de mutaciones germinales en todos los pacientes diagnosticados de PPGL1, se desconoce en qué medida esta recomendación se está llevando a la práctica en el medio clínico. El principal objetivo de nuestro trabajo fue determinar en cuántos de los pacientes diagnosticados de feocromocitoma o paraganglioma se había investigado la presencia de mutaciones germinales predisponentes y, en segundo lugar, qué mutaciones se encontraban en los pacientes estudiados. Decidimos incluir a los pacientes diagnosticados a partir del 2010 porque en ese momento ya se conocían los principales genes predisponentes a PPGL, se había documentado el porcentaje significativo de pacientes no sindrómicos y sin antecedentes familiares que presentaban mutaciones en dichos genes por parte de varios grupos, y algunos expertos ya recomendaban que se estudiaran estas mutaciones en todos o en la mayor parte de los pacientes con PPGL9,11,14. No recogimos información acerca del estudio de mutaciones somáticas en tejido tumoral, ya que las recomendaciones acerca de su realización son aún vagas en las guías de práctica clínica de los últimos años.
El estudio de mutaciones germinales predisponentes había sido realizado en el 54% de los pacientes diagnosticados de PPGL. No se han realizado estudios similares con los que comparar esta cifra ni en nuestro país ni en otros, pero la variabilidad que hemos encontrado entre distintas especialidades en un mismo hospital nos permite aventurar que probablemente haya mucha variabilidad entre distintos entornos clínicos. Nuestro centro de trabajo es un hospital de tercer nivel, universitario, pero no especializado en esta patología y, por lo tanto, probablemente representativo de la mayoría de los centros hospitalarios donde los pacientes con PPGL de nuestro país son atendidos.
La probabilidad de encontrar mutaciones en línea germinal en un determinado paciente es mayor cuando este es más joven, tiene antecedentes familiares o tumores característicos de los síndromes asociados a PPGL, cuando su PPGL es extraadrenal, bilateral, múltiple o metastásico2,3. Pudimos comprobar que estos factores de riesgo se habían tenido en cuenta a la hora de solicitar el estudio genético en nuestro medio, ya que los pacientes con estudio genético eran más jóvenes, tenían más cuadros sindrómicos y más antecedentes familiares, y tenían con mayor frecuencia tumores múltiples. De hecho, todos los pacientes con antecedentes familiares o con características clínicas de síndrome MEN2A habían sido estudiados. También la única paciente con PPGL maligno había sido estudiada sin encontrar mutación germinal. Sin embargo, la localización extraadrenal del tumor no se asoció a una mayor solicitud de estudios genéticos, especialmente en los tumores únicos de cabeza y cuello. Por otro lado, llama la atención que la probabilidad de solicitud del estudio genético depende también en gran medida del servicio médico encargado de atender al paciente.
El porcentaje de pacientes con mutaciones (33%) de nuestra serie es similar a los comunicados por otros grupos y también la distribución de los genes mutados es similar a la de otras series5,6,8-14. Es posible que hubiéramos encontrado un porcentaje diferente si se hubieran estudiado todos los pacientes: por una parte, los no estudiados tenían mayor edad y menos cuadros sindrómicos, menos antecedentes familiares y menos tumores múltiples, lo que hace más improbable la presencia de mutaciones germinales, pero, por otro lado, 10 de los 18 pacientes no estudiados tenían tumores extraadrenales, que con frecuencia se asocian a mutaciones.
El grupo de pacientes con menor probabilidad de tener una mutación germinal son aquellos con un feocromocitoma suprarrenal unilateral no metastásico, no sindrómico y sin antecedentes familiares, sobre todo si son mayores de 45 años. En nuestra serie, se realizó estudio genético a 7 de los 17 pacientes con estas características, todos ellos mayores de 45 años, y no encontramos mutaciones germinales en ninguno de los 7. Otros grupos han publicado porcentajes de mutación entre el 4 y el 28% en estos pacientes: Cascón et al. encontraron mutaciones en un 3,9% de los pacientes no sindrómicos, sin antecedentes familiares y mayores de 40 años9, y Currás-Freixes et al. en el 4,5% de los feocromocitomas únicos, esporádicos, no sindrómicos10. Un porcentaje algo superior se encuentra en la serie de Jafri et al., que describen mutaciones en el 28% de los feocromocitomas esporádicos, pero la edad media era de 36 años, claramente menor que en las otras series13.
Entre las limitaciones de nuestro estudio, cabe la posibilidad de que algún paciente haya rechazado el estudio genético y que esto no se haya recogido en su historia clínica, algo poco probable. Por otro lado, la revisión de las historias clínicas sugería que la anamnesis no siempre era exhaustiva en la búsqueda de antecedentes familiares (interrogando acerca de PPGL en localizaciones distintas de la del paciente y de otros tumores sindrómicos). También hubo variabilidad en el número de genes analizados en los pacientes con estudio genético negativo. Es poco probable que la utilización de secuenciación clásica en la mayoría de los pacientes y de técnicas NGS en los más recientes haya influido en los resultados globales.
El conocimiento de las bases moleculares de la patogenia de feocromocitomas y paragangliomas crece a pasos de gigante en los últimos años, permitiendo vaticinar que en un futuro cercano identificar la mutación de un determinado paciente contribuirá a elegir el tratamiento más adecuado y a conocer el pronóstico de cada caso. Nuestro estudio pone de manifiesto una clara variabilidad entre distintos servicios en la probabilidad de solicitud de estudio genético y una injustificada baja solicitud en el caso de paragangliomas únicos de cabeza y cuello, por lo que consideramos necesario establecer protocolos de atención a estos pacientes consensuados entre los distintos especialistas implicados.
AutoríaCompetencias: CMJ y MO, realizaron la recogida de datos de las historias clínicas. RPQ, CG, PP y CL fueron los médicos que atendieron a los pacientes en el servicio de endocrinología. A CMJ y CL corresponde la idea original del trabajo, la redacción y corrección final del manuscrito.
Conflicto de interesesLos autores de este trabajo declaran no tener ningún conflicto de interés. Este trabajo es original y no ha sido publicado con anterioridad. Todos los autores aprueban su publicación.
Al grupo de Cáncer endocrino hereditario del CNIO y en particular a María Currás y Mercedes Robledo, por la realización de los estudios genéticos.
