




La mayoría de los feocromocitomas y paragangliomas son esporádicos. Sin embargo, hasta un 40% de ellos tienen un origen hereditario, debido a mutaciones en línea germinal en alguno de los hasta la fecha 17 genes conocidos como causantes de estos tumores1. De entre todos ellos, los genes de las diferentes subunidades de la succinato deshidrogenasa y el gen VHL son los más frecuentemente afectados2.
La fumarato hidratasa es una enzima que participa en el ciclo de Krebs, catalizando la conversión de fumarato en malato. Las mutaciones inactivantes en el gen que la codifica (FH) conllevan un incremento de los niveles intracelulares de fumarato, conduciendo a la activación de la vía de la pseudohipoxia y la transcripción de diferentes genes implicados en angiogénesis y crecimiento tumoral3. Las mutaciones heterocigotas en línea germinal del gen FH se han asociado previamente al síndrome de leiomiomatosis y cáncer renal hereditario4 (HLRCC, por sus siglas en inglés, OMIM # 150800), una patología de herencia autosómica dominante, caracterizada por el desarrollo de múltiples leiomiomas uterinos y cutáneos y carcinoma papilar renal de tipo 2, una agresiva neoplasia renal de mal pronóstico. En el año 2013, sin embargo, se identificaron por primera vez mutaciones de FH en algunos pacientes con feocromocitoma y paraganglioma hereditario, con una aparente elevada predisposición para desarrollar enfermedad metastásica5–7. Hasta la fecha, ninguno de los casos descritos en la literatura había combinado feocromocitomas o paragangliomas con HLRCC. Presentamos aquí el primer caso.
Se trata de una mujer que se presentó en abril de 2007, con 44 años edad. Su historia previa incluía una histerectomía por leiomiomas uterinos y tenía también antecedentes familiares de miomatosis uterina en su madre. Consultó debido a la aparición de episodios, cada vez más frecuentes, de palpitaciones, cefalea, rubor facial y mareos, asociados a una elevación leve de la presión arterial. El estudio hormonal mostró normetanefrina en plasma>1.000 pg/ml (valor normal [VN]:<180 pg/ml), normetanefrina en orina de 24 horas de 8045μg/24 horas (VN: 88–444μg/24 horas) y cromogranina A de 1074 ng/mL (VN: 19,4– 98,1 ng/ml), siendo los valores de metanefrinas normales en plasma y orina. Una TAC abdominal reveló una masa quística en el polo inferior del riñón izquierdo, de 13 x 13 x 9,6cm de diámetro, con múltiples engrosamientos nodulares de su pared, hiperintensos tras la administración de contraste (quiste grado III de Bosniak). En la glándula suprarrenal ipsilateral se observó una masa con un borde nodular hipercaptante, con paredes gruesas e irregulares, y una gran zona central quística, que se interpretó como compatible, en primer lugar, con un feocromocitoma. Previa preparación prequirúrgica con doxazosina durante tres semanas, y propranolol durante los últimos 10 días, la paciente se intervino mediante nefrectomía y adrenalectomía izquierdas, sin complicaciones. La anatomía patológica confirmó el diagnóstico de un feocromocitoma de 9,5cm de diámetro, con focos de necrosis y extensas áreas de hialinización. El índice mitótico era bajo, no había pleomorfismo nuclear y no se identificó invasión vascular. El tumor presentaba una puntuación de 2, indicando bajo riesgo de metástasis, en la escala Pheochromocytoma of the Adrenal gland Scaled Score, a expensas de necrosis confluente o de tipo comedo, y una puntuación de 2 en la escala Grading of Adrenal and extra-adrenal Pheochromocytomas and relationship to Prognosis, que correspondería a un tumor bien diferenciado, aunque no se pudo medir la expresión de Ki-67. Así mismo, en la pieza de nefrectomía se identificó un carcinoma de células renales grado 4 según la clasificación de Fuhrman, con extensa degeneración quística y con patrón túbulo-papilar, dispuesto en múltiples nódulos en la pared de un quiste de 10cm de dimensión mayor, que se extendía focalmente por fuera de la cápsula, infiltrando el tejido adiposo perirrenal. Tras la cirugía se normalizaron los valores de normetanefrina y noradrenalina en plasma y en orina y se resolvió el cuadro clínico de la paciente. Desde ese momento, ha mantenido seguimiento periódico con estudios repetidos de TAC de abdomen, gammagrafía-SPECT con meta-I-bencil-guanidina y determinaciones de normetanefrina y metanefrina en orina de 24 horas, que han descartado recidivas locales y metástasis.
Un estudio inicial en el que se secuenciaron los genes VHL, SDHB y RET, fue negativo. En noviembre de 2015 se hizo una ampliación del estudio genético mediante el análisis de un panel de genes asociados a feocromocitoma hereditario (VHL, RET, EPAS1/HIF2, HRAS, TMEM127, MAX, SDHA, SDHB, SDHC, SDHAF2, FH, MDH2, EGLN1 y NF1)8, en el que detectó una mutación que afectaba un sitio canónico de splicing en el exón 4 del gen FH (NM_000143.3:c.555+1G>A). Dicha mutación había sido previamente descrita en una familia española con leiomiomatosis cutánea y uterina9 y en un caso de una serie de pacientes con HLRCC en Francia10, pero no en pacientes con feocromocitomas o paragangliomas.
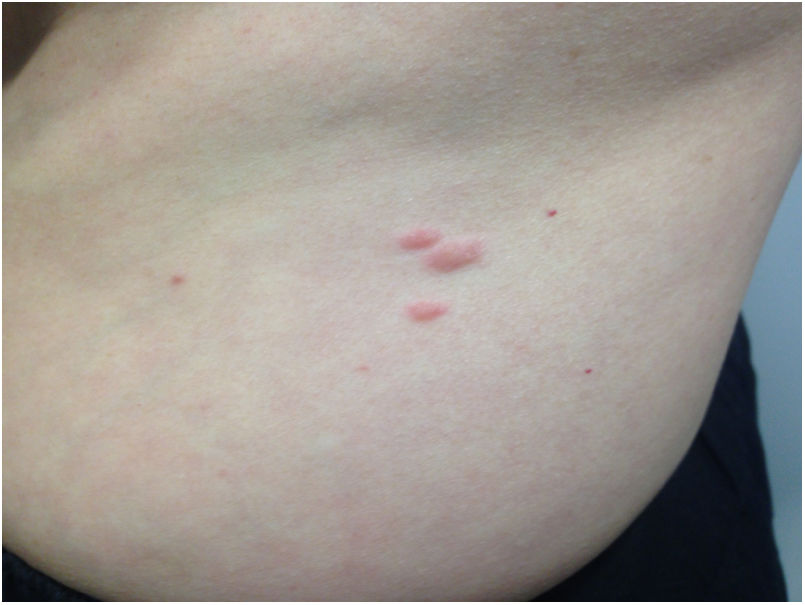
Tras conocer estos resultados, en una exploración física más detallada se comprobó la existencia de pápulas rosadas en antebrazos y tronco, de 0,5 a 2cm de diámetro, que la paciente había notado desde hacía 2 años (fig. 1). La biopsia de una de estas lesiones confirmó que se trataba de leiomiomas cutáneos.

El estudio genético de la mutación en las dos hijas y una hermana de la paciente fue negativo.
Las mutaciones en el gen FH son responsables del síndrome HLRCC, pero constituyen también una causa excepcional de feocromocitoma/paraganglioma hereditario. En nuestro conocimiento, solo se han descrito siete casos de pacientes portadores de una mutación en el gen FH con feocromocitoma y/o paranganglioma5–7, tres de ellos con tumores metastásicos. Ninguno de estos casos presentaba leiomiomatosis cutánea ni cáncer renal y solo uno5 tenía antecedentes de leiomiomatosis uterina. El presente caso demuestra que los sujetos que presentan mutaciones patogénicas en heterocigosis en el gen FH están en riesgo de desarrollar tanto feocromocitomas/paragangliomas como síndrome HLRCC, y que debería procederse al cribado de ambos trastornos una vez se haya establecido el diagnóstico genético.
Los autores desean hacer constar su agradecimiento a la Dra. Mercedes Robledo, (Centro Nacional de Investigaciones Oncológicas, Madrid), por su revisión del manuscrito.
