





Hace más de cincuenta años que empezaron los ensayos clínicos aleatorizados sobre los efectos cardiovasculares de los fármacos hipoglucemiantes en personas con lo que hoy se conoce como diabetes mellitus tipo2. Este proceso se ha acelerado en la última década con la aparición de nuevas categorías de fármacos hipoglucemiantes, lo que ha llevado al desarrollo de ensayos clínicos aleatorizados para evaluar su seguridad cardiovascular. Los denominados CardiovascularOutcomeTrials han proporcionado una ingente cantidad de información que debería analizarse críticamente para poderla transformar en conocimiento aplicable a la práctica clínica. Para ello, tras comentar las guías a las que se han adherido esos ensayos, se comentan una serie de conceptos para interpretarlos mejor (desde los tipos de análisis hasta la definición de objetivos y la evaluación crítica de los resultados), y se finaliza con una mención a las nuevas guías a las que se adherirán los futuros ensayos que se diseñen para evaluar la seguridad de los nuevos fármacos hipoglucemiantes.
Randomized clinical trials on the cardiovascular effects of hypoglycemic drugs on people with type2 diabetes mellitus began more than fifty years ago. In the last decade, the emergence of new classes of hypoglycemic drugs has led to the development of randomized clinical trials to assess their cardiovascular safety. Known as Cardiovascular Outcome Trials, they have provided a lot of new information that needs to be critically appraised if the knowledge obtained is to be applicable in clinical practice. To this end, the current article first comments on the guidelines to which these trials have adhered, then reviews some concepts for improving their interpretation (such as different types of analyses, the definition of objectives and the evaluation of their results), and concludes by mentioning the new guidelines to which future trials designed to evaluate the safety of new hypoglycemic drugs should adhere.
El primer ensayo clínico aleatorizado (ECA) sobre efectos cardiovasculares de fármacos hipoglucemiantes en lo que hoy se consideraría una diabetes tipo2 (DM2) se inició en la década de los sesenta del pasado siglo. Era el UniversityGroupDiabetesProgram, y sus resultados sugirieron que la insulina era más segura que la tolbutamida o la fenformina, aunque generaron un intenso debate durante años1. A finales de la década de los setenta del siglo pasado en el Reino Unido se inició el UnitedKingdomProspective Diabetes Study (UKPDS). Este estudio reclutó 4.209 personas con DM2 de inicio reciente, dándose por finalizado (en una primera fase) en 1997. Desde el punto de vista cardiovascular concluyó que para un objetivo formado por una miscelánea de 12 componentes (muerte súbita, muerte por hipo o hiperglucemia, infarto de miocardio [IM] fatal o no fatal, angina, insuficiencia cardíaca, accidente cerebrovascular [ACV], fallo renal, amputación de al menos un dedo, hemorragia vítrea, retinopatía tratada con láser, amaurosis de un ojo o extracción de cataratas), un control glucémico más intensivo con una HbA1c media del 7,0% era mejor que uno estándar con HbA1c media del 7,8%, con un riesgo relativo (RR) del 0,88 (IC95%: 0,78-0,99)2. En un contexto de absoluta falta de este tipo de estudios señalamos estas limitaciones:
- 1.
Largo período de reclutamiento (de 1977 a 1991) con repetidas modificaciones del diseño inicial3.
- 2.
Los criterios diagnósticos de DM2 de la época exigían mayores valores de glucemia que los actuales.
- 3.
La HbA1c no estuvo disponible los primeros años.
- 4.
El objetivo compuesto comentado incluyó eventos de procedimiento —p.ej., extracción de cataratas— cuya adjudicación dependía del centro participante.
- 5.
El límite superior del IC95% del resultado sobre ese objetivo fue del 0,99, es decir, en el límite de la significación estadística.
- 6.
En el análisis se hicieron decenas de comparaciones sin ajustar el valor de p2.
- 7.
En la evaluación de los componentes de ese objetivo compuesto no se consideró el momento en que aparecían los eventos, solo si estos habían o no aparecido al cierre del estudio, por lo que los resultados se dieron como RR (ver siguiente apartado).
- 8.
De los resultados en las 342 personas con sobrepeso que recibieron metformina, se infirió que esta tenía un efecto cardiovascular beneficioso4, hecho aún por demostrar.
En ese momento no se detectó beneficio del tratamiento intensivo sobre un objetivo compuesto de IM fatal o no fatal y muerte súbita (p=0,052), pero sí se reportó 10años después para IM, especialmente para los que recibieron metformina (p=0,005)5. Resumiendo, el UKPDS no mostró un efecto cardiovascular beneficioso de la intensificación del tratamiento de la hiperglucemia, como tampoco lo hicieron otros tres ECA multicéntricos6-8 que le siguieron en la primera década de este siglo. De hecho, uno de ellos tuvo que cancelarse prematuramente por incremento de la mortalidad total [MT] en el grupo con tratamiento intensivo6, lo que no fue óbice para que se incluyera junto con los otros tres en un metaanálisis que concluyó que un control intensivo de la glucemia se asociaba a una hazardratio (HR) de 0,91 (IC95%: 0,78-0,99) para un objetivo compuesto de muerte cardiovascular [MCV], IM no fatal y ACV no fatal, conocido posteriormente como majoradversecardiovascularevents-3 (MACE-3)9. Aquí, de nuevo, el resultado estuvo en el límite de la significación estadística.
Dudas sobre la seguridad cardiovascular de las glitazonasTras la comercialización de las primeras glitazonas, el ECA PROspective pioglitAzone Clinical Trial In macroVascular Events (PROactive) no mostró beneficio de pioglitazona frente a placebo en su objetivo primario [OP] (MT, IM no fatal —incluido silentes—, ACV no fatal, síndrome coronario agudo, revascularización y amputación), pero sí lo hizo en un objetivo secundario que solo incluía los tres primeros componentes del primario excluyendo los infartos silentes (HR: 0,84; IC95%: 0,72-0,98)10. Se concluyó que el OP no había sido significativo por incluir eventos de procedimiento (p.ej., revascularización) que podían haberse indicado de forma diferente en los distintos centros participantes. Tras no conseguir el muraglitazar ser comercializado por sus efectos adversos cardiovasculares deletéreos11, un metaanálisis con 42ECA de al menos 24semanas de duración mostró una oddsratio (OR) de 1,43 (IC95%: 1,03-1,98) para IM en contra de la rosiglitazona12. Esto causó una fuerte polémica, la retirada del mercado de la rosiglitazona y que en 2008 la FoodDrugAdministration (FDA) emitiera una guía sobre cómo evaluar el riesgo cardiovascular de los nuevos fármacos hipoglucemiantes (NFH) para el tratamiento de la DM2. Esta guía puede resumirse así13,14:
- 1.
La evaluación del riesgo cardiovascular debería incluir como mínimo un OP que incluya el estudio de los tres componentes del MACE-3 (MCV, IM y ACV), aunque puedan incluirse también otros eventos, como hospitalización por síndrome coronario agudo u hospitalización por insuficiencia cardíaca (HIC), o la revascularización urgente.
- 2.
La definición de todos esos eventos debería preespecificarse, y su adjudicación deberá realizarla un comité independiente.
- 3.
Esa evaluación requiere que el programa de desarrollo del NFH tenga ECA de fase2 y fase3 de suficiente tamaño y duración como para que su metaanálisis pueda responder adecuadamente a las recomendaciones de la guía. En lugar de esto, puede hacerse un ECA específico para evaluar el riesgo cardiovascular del NFH, conocido como un CardiovascularOutcomeTrial (CVOT). En la práctica todos los NFH han presentado su propio CVOT.
- 4.
Las nuevas insulinas quedan exentas de adherirse a estas recomendaciones.
- 5.
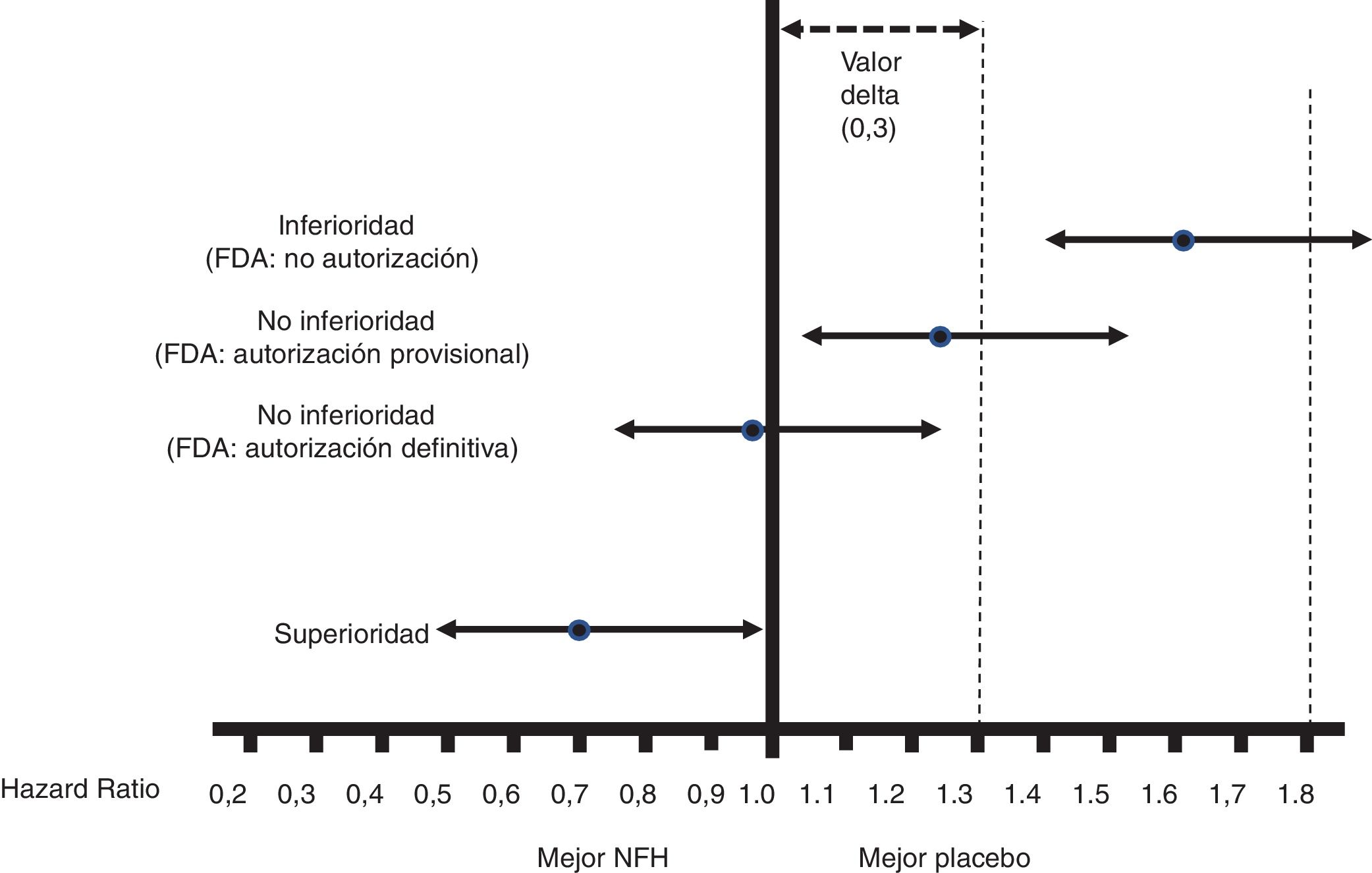
La interpretación de los resultados del OP de esos CVOT aparece en la figura 1.
La aceptación de un valor delta <0,3 para aceptar que un NFH es seguro (o no inferior). 2)Atender al límite superior del IC95% (p bilateral) de la diferencia entre las personas con el NFH y las que no lo recibieron, en relación al OP (en general MACE-3): Si es ≥1,8, el NFH no se autorizará por su elevado riesgo cardiovascular; si es <1,3, se asumirá que es seguro y se autorizará su comercialización; si es ≥1,3 pero <1,8, se autorizará provisionalmente su comercialización (o no se suspenderá), a la espera de más resultados que confirmen que ese límite superior de la diferencia es <1,3. La guía no recomienda que el NFH muestre beneficio cardiovascular, es decir, que ese límite superior de la diferencia sea <1,0 (superioridad); solo recomienda acreditar no inferioridad (punto de corte, <1,3).") Figura 1.
Figura 1.Interpretación de los resultados sobre el OP de los CVOT según la guía de la FDA de 200813. Esta interpretación se basa en: 1)La aceptación de un valor delta <0,3 para aceptar que un NFH es seguro (o no inferior). 2)Atender al límite superior del IC95% (p bilateral) de la diferencia entre las personas con el NFH y las que no lo recibieron, en relación al OP (en general MACE-3): Si es ≥1,8, el NFH no se autorizará por su elevado riesgo cardiovascular; si es <1,3, se asumirá que es seguro y se autorizará su comercialización; si es ≥1,3 pero <1,8, se autorizará provisionalmente su comercialización (o no se suspenderá), a la espera de más resultados que confirmen que ese límite superior de la diferencia es <1,3. La guía no recomienda que el NFH muestre beneficio cardiovascular, es decir, que ese límite superior de la diferencia sea <1,0 (superioridad); solo recomienda acreditar no inferioridad (punto de corte, <1,3).
(0.1MB).
La aceptación de un valor delta <0,3 para aceptar que un NFH es seguro (o no inferior). 2)Atender al límite superior del IC95% (p bilateral) de la diferencia entre las personas con el NFH y las que no lo recibieron, en relación al OP (en general MACE-3): Si es ≥1,8, el NFH no se autorizará por su elevado riesgo cardiovascular; si es <1,3, se asumirá que es seguro y se autorizará su comercialización; si es ≥1,3 pero <1,8, se autorizará provisionalmente su comercialización (o no se suspenderá), a la espera de más resultados que confirmen que ese límite superior de la diferencia es <1,3. La guía no recomienda que el NFH muestre beneficio cardiovascular, es decir, que ese límite superior de la diferencia sea <1,0 (superioridad); solo recomienda acreditar no inferioridad (punto de corte, <1,3).")
Todos los CVOT presentados desde la emisión de esta guía han cumplido el criterio de no inferioridad de <1,3. En ocasiones, como ha ocurrido con empagliflozina, canagliflozina o semaglutida (subcutáneo u oral), el ECA se ha diseñado para pasar el punto de corte de 1,8, aumentado luego el seguimiento (empagliflozina)15 o el tamaño muestral (canagliflozina)16 para pasar el punto de corte de <1,3, o simplemente no haciendo nada adicional por haber pasado ya el punto de corte de <1,0 con el diseño inicial (semaglutida)17.
Interpretación de los CVOTHasta el 5 de agosto de 2020 se han publicado 15 CVOT. En todos ellos el comparador ha sido un placebo, salvo en uno, que fue otro hipoglucemiante, y no se analizará18, como tampoco se incluirán los realizados tras un evento coronario agudo19,20, o con NFH aún no comercializados en nuestro país21. En las tablas 1 y 2 aparecen los 11CVOT restantes que analizaremos: tres con inhibidores del co-transportador sodio-glucosa tipo2 (ISGLT2)15,16,22, cinco con análogos del receptor del glucagon-like peptide 1 (ARGLP1)17,23-26 y tres con inhibidores de la dipeptidil-peptidasa 4 (IDDP4)27-29. En ellos, vamos a considerar los siguientes puntos:
Características de los CVOT analizados
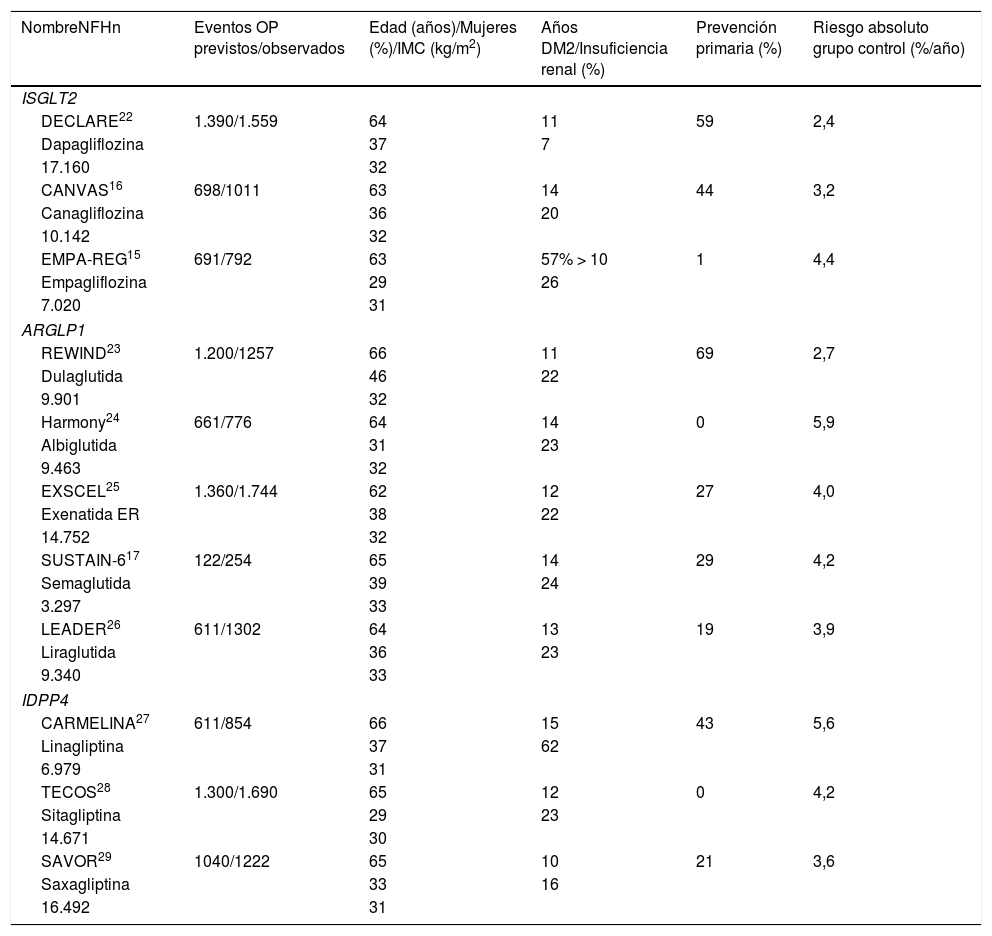
| NombreNFHn | Eventos OP previstos/observados | Edad (años)/Mujeres (%)/IMC (kg/m2) | Años DM2/Insuficiencia renal (%) | Prevención primaria (%) | Riesgo absoluto grupo control (%/año) |
|---|---|---|---|---|---|
| ISGLT2 | |||||
| DECLARE22 | 1.390/1.559 | 64 | 11 | 59 | 2,4 |
| Dapagliflozina | 37 | 7 | |||
| 17.160 | 32 | ||||
| CANVAS16 | 698/1011 | 63 | 14 | 44 | 3,2 |
| Canagliflozina | 36 | 20 | |||
| 10.142 | 32 | ||||
| EMPA-REG15 | 691/792 | 63 | 57% > 10 | 1 | 4,4 |
| Empagliflozina | 29 | 26 | |||
| 7.020 | 31 | ||||
| ARGLP1 | |||||
| REWIND23 | 1.200/1257 | 66 | 11 | 69 | 2,7 |
| Dulaglutida | 46 | 22 | |||
| 9.901 | 32 | ||||
| Harmony24 | 661/776 | 64 | 14 | 0 | 5,9 |
| Albiglutida | 31 | 23 | |||
| 9.463 | 32 | ||||
| EXSCEL25 | 1.360/1.744 | 62 | 12 | 27 | 4,0 |
| Exenatida ER | 38 | 22 | |||
| 14.752 | 32 | ||||
| SUSTAIN-617 | 122/254 | 65 | 14 | 29 | 4,2 |
| Semaglutida | 39 | 24 | |||
| 3.297 | 33 | ||||
| LEADER26 | 611/1302 | 64 | 13 | 19 | 3,9 |
| Liraglutida | 36 | 23 | |||
| 9.340 | 33 | ||||
| IDPP4 | |||||
| CARMELINA27 | 611/854 | 66 | 15 | 43 | 5,6 |
| Linagliptina | 37 | 62 | |||
| 6.979 | 31 | ||||
| TECOS28 | 1.300/1.690 | 65 | 12 | 0 | 4,2 |
| Sitagliptina | 29 | 23 | |||
| 14.671 | 30 | ||||
| SAVOR29 | 1040/1222 | 65 | 10 | 21 | 3,6 |
| Saxagliptina | 33 | 16 | |||
| 16.492 | 31 | ||||
ARGLP1: análogos del receptor del GLP1; CARMELINA: Cardiovascular and Renal Microvascular outcome study with LINAgliptin; DM2: diabetes mellitus tipo2; IDPP4: inhibidores de la dipeptidil-peptidasa 4; ISGLT2: inhibidores del co-transportador sodio-glucosa2; NFH: nuevo fármaco hipoglucemiante; OP: objetivo primario.
Características de los CVOT analizados
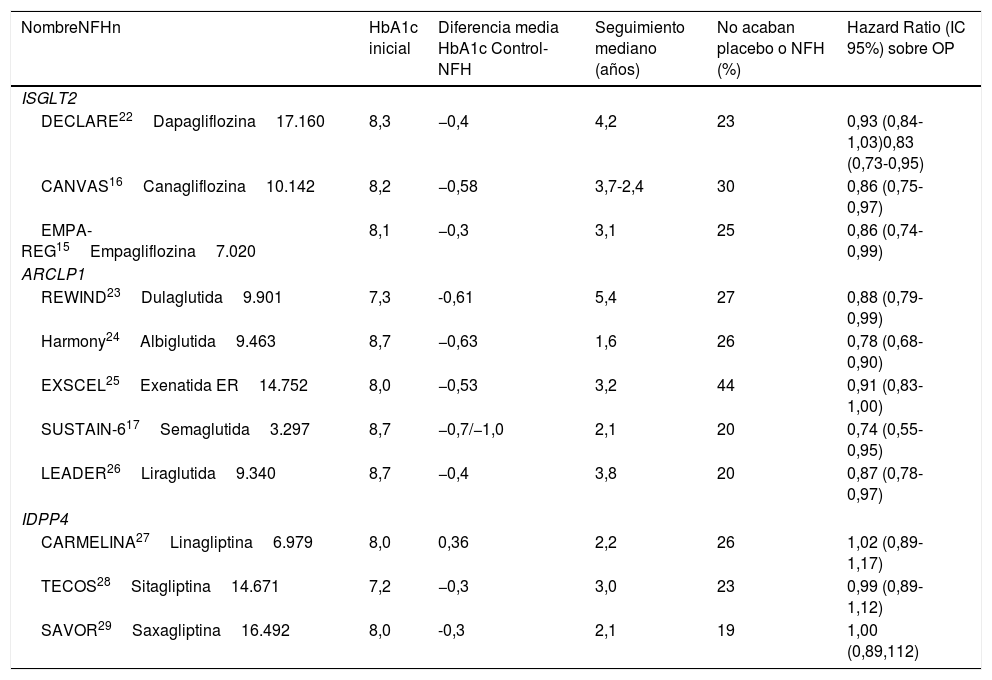
| NombreNFHn | HbA1c inicial | Diferencia media HbA1c Control-NFH | Seguimiento mediano (años) | No acaban placebo o NFH (%) | Hazard Ratio (IC 95%) sobre OP |
|---|---|---|---|---|---|
| ISGLT2 | |||||
| DECLARE22Dapagliflozina17.160 | 8,3 | −0,4 | 4,2 | 23 | 0,93 (0,84-1,03)0,83 (0,73-0,95) |
| CANVAS16Canagliflozina10.142 | 8,2 | −0,58 | 3,7-2,4 | 30 | 0,86 (0,75-0,97) |
| EMPA-REG15Empagliflozina7.020 | 8,1 | −0,3 | 3,1 | 25 | 0,86 (0,74-0,99) |
| ARCLP1 | |||||
| REWIND23Dulaglutida9.901 | 7,3 | -0,61 | 5,4 | 27 | 0,88 (0,79-0,99) |
| Harmony24Albiglutida9.463 | 8,7 | −0,63 | 1,6 | 26 | 0,78 (0,68-0,90) |
| EXSCEL25Exenatida ER14.752 | 8,0 | −0,53 | 3,2 | 44 | 0,91 (0,83-1,00) |
| SUSTAIN-617Semaglutida3.297 | 8,7 | −0,7/−1,0 | 2,1 | 20 | 0,74 (0,55-0,95) |
| LEADER26Liraglutida9.340 | 8,7 | −0,4 | 3,8 | 20 | 0,87 (0,78-0,97) |
| IDPP4 | |||||
| CARMELINA27Linagliptina6.979 | 8,0 | 0,36 | 2,2 | 26 | 1,02 (0,89-1,17) |
| TECOS28Sitagliptina14.671 | 7,2 | −0,3 | 3,0 | 23 | 0,99 (0,89-1,12) |
| SAVOR29Saxagliptina16.492 | 8,0 | -0,3 | 2,1 | 19 | 1,00 (0,89,112) |
ARGLP1: análogos del receptor del GLP1; CARMELINA: Cardiovascular and Renal Microvascular outcome study with LINAgliptin; IDPP4: inhibidores de la dipeptidil-peptidasa 4; ISGLT2: inhibidores del co-transportador sodio-glucosa 2; NFH: nuevo fármaco hipoglucemiante; OP: objetivo primario.
Cuando se diseña el CVOT, y al menos antes de tener ningún resultado, se debe dar a conocer su plan de análisis estadístico, donde se asumen unos valores de p (significación estadística) proporcionales al grado de incertidumbre de ese momento. Esto ha ocurrido en todos los CVOT para su OP. Cuando ya se conocen los resultados pueden hacerse análisis post hoc de los mismos, pero sus resultados siempre serán exploratorios y generadores de hipótesis, pues se basan en el comportamiento ya observado de los resultados ya reportados.
2. No inferioridad frente a superioridad30,31Todos los CVOT son ECA de no inferioridad que se adhieren a la guía de la FDA de 2008, salvo el Researching cardiovascular Events with a Weekly INcretin in Diabetes (REWIND), que se planteó de inicio como de superioridad23. A diferencia de los ECA de superioridad, en los de no inferioridad se define un valor delta que es la máxima diferencia aceptable entre el NFH y su comparador en el OP (casi siempre: MACE-3) para considerar que el NFH no es inferior. La guía de la FDA estableció ese valor delta en <0,3, y por eso el punto de corte para definir un NFH como seguro fue de <1,3 (fig. 1). La no inferioridad no implica equivalencia, pero sí intercambiabilidad. Además, no puede establecerse con un ECA para demostrar superioridad de un NFH frente a su comparador, cuando no se demuestra tal superioridad, ni tampoco con múltiples ECA de pequeño tamaño (p.ej., n<800) que no muestren diferencias entre NFH y comparador en un metaanálisis. Los ECA de no inferioridad precisan mayor tamaño muestral que los de superioridad (ver punto4), por lo que dentro de un mismo CVOT es admisible evaluar primero la no inferioridad y, si esta se confirma, evaluar luego la superioridad, siempre que todo ello sea preespecificado, hecho común en los CVOT analizados. Si en un ECA de superioridad esta no se demuestra, no puede evaluarse luego una posible no inferioridad, ya que requeriría mayor tamaño muestral.
3. Análisis por protocolo (perprotocol o astreated) o por intención de tratar (intention-to-treat)32En el análisis por protocolo solo se evalúan los participantes que cumplieron todo el tratamiento asignado, fuera NFH o comparador. Este número siempre es menor al de la población por intención de tratar, ya que esta incluye a todos los participantes aleatorizados a recibir NFH o comparador, lo hayan cumplido completamente o no. El análisis por intención de tratar diluiría el potencial efecto del NFH, favoreciendo la no inferioridad. Según Pocock et al.32, en los CVOT se deberían realizar los dos tipos de análisis para comprobar su congruencia.
4. Tamaño muestralAl evaluar un NFH frente a un comparador se definen dos tipos de errores: a)Error tipoI (alfa): el NFH aumentaría el riesgo cardiovascular, pero realmente no lo hace (falso positivo). El valor de alfa en los CVOT es de 0,05 bilateral. b)Error tipoII (beta): el NFH no aumentaría el riesgo cardiovascular, pero realmente sí lo hace (falso negativo). La potencia es (1−beta)×100, y suele ser del 90% en los CVOT.
En los ECA de no inferioridad, cinco factores aumentan su tamaño muestral:33 a)Una menor tasa de eventos del OP en el grupo control. b)Una menor tasa de eventos en el OP con el NFH (menor efecto del NFH sobre el OP). c)Un menor riesgo alfa. d)Un menor valor delta; la FDA recomendó un valor delta de <0,3 (lo que implicaba reconocer como seguros NFH que podían aumentar el riesgo de evento del OP hasta en un <30%) de forma pragmática, porque por ejemplo para valores delta de <0,1 se habrían necesitado CVOT con más de 50.000 participantes. e)Una mayor potencia estadística; esta depende del número de eventos esperados en el OP, que para una HR de 1,3 suele ser >700. Los CVOT se plantean de forma que se detienen cuando se alcanza un número preespecificado de esos eventos (se dice que son event-driven trials). Todos los CVOT analizados superaron ese número (tabla 1).
5. Criterios de inclusión/exclusiónLos criterios de inclusión/exclusión determinan la validez externa del CVOT (extrapolación de sus resultados a todas las personas con DM2). Aparte de las características demográficas de los participantes, un criterio muy importante es el porcentaje de participantes que aún no han sufrido eventos cardiovasculares (prevención primaria), pues tienen un menor riesgo absoluto (RA) de sufrirlos que los que ya los tuvieron antes (prevención secundaría) (tabla 1). Al comparar los CVOT hay que considerar que los criterios para elegir los participantes en prevención primaria no suelen ser iguales. Así, por ejemplo, en el CANagliflozin cardiovascular Assessment Study (CANVAS)16 el grupo en prevención primaria presentaba mayor RA de MACE-3 por sus características clínicas que el de prevención primaria del Dapagliflozin Effect on CardiovascuLAR Events-TIMI 58 (DECLARE)22, y tampoco son comparables los criterios del REWIND23 con los del Trial to Evaluate Cardiovascular and Other Long-term Outcomes with Semagluide in Subjects with Type2 Diabetes (SUSTAIN-6)17o el Liraglutide Effect and Action in Diabetes: Evaluation of cardiovascular outcome Results (LEADER)26. Además, los primeros eventos cardiovasculares pueden ser asintomáticos. Por todo ello, parece que lo más razonable al comparar los CVOT sería hacerlo según el RA del OP en el grupo control34. Así, el DECLARE22 fue el CVOT con menor RA (2,4%/año o 2,4 por 100 pacientes-año) y el Harmony Outcome (Harmony) el que lo tuvo mayor (5,9%/año) (tabla 1)24.
Al comparar los grupos resulta interesante observar la evolución temporal del porcentaje acumulado de participantes con algún evento del OP con las curvas de Kaplan-Meier. Así, en el Empagliflozin Cardiovascular Outcome Event Trial in Type2 diabetes Mellitus Patients (EMPA-REG) se empezaron a separar en cuestión de meses a favor de su NFH15, mientras que lo hicieron casi a los 2años en el LEADER26. Esto llevó a sugerir que el NFH del primer estudio tendría un efecto beneficioso por un mecanismo más hemodinámico, mientras que el del segundo lo tendría por un efecto más antiarteriosclerótico.
6. Período de run-inEs un período previo a la aleatorización cuyo objetivo es asegurar que los participantes que se vayan a aleatorizar tienen buena adherencia a los tratamientos del ensayo, aun a costa de disminuir la validez externa del mismo. La mejor manera de evaluarlo es observando el gráfico de flujo de participantes que aparece en estas publicaciones. Conviene entonces evaluar tanto el porcentaje de potenciales participantes que no superaron la fase del run-in como el motivo por el que ello sucedió, aunque a veces este no se especifique en un elevado número de casos23.
7. Intervenciones, aleatorización y enmascaramientoEn todos los CVOT la intervención consistió en añadir a los participantes aleatoriamente a su tratamiento habitual para la DM2 el NFH o su placebo, minimizando las diferencias de control glucémico entre grupos, es decir, buscando la glycemicequipoise. Esto es así porque se trata de evaluar la seguridad cardiovascular del NFH independientemente de su efecto hipoglucemiante. Para conseguir este equilibrio se suelen utilizar otros fármacos hipoglucemiantes, cuyo uso no suele ser autorizado hasta varias semanas/meses después de la aleatorización. Esto determina que el valor medio de HbA1c siempre sea algo menor en los participantes con el NFH (tabla 2). En algunos CVOT se han usado dos dosis distintas del NFH, pero preespecificándose que para evaluar el OP esas dosis se considerarían conjuntamente15-17. El CVOT con mayor diferencia de HbA1c entre grupos fue el SUSTAIN-617, congruente con el mayor efecto sobre la HbA1c de su NFH frente a todos los otros hipoglucemiantes a los que se ha comparado hasta ahora35-37.
Todos los CVOT han tenido otras tres características33: a)El investigador desconocía de antemano el tratamiento que se asignaría al participante caso de ser incluido (allocation concealment). b)Como mínimo, tanto investigador como participante desconocían la naturaleza del producto recibido, fuera NFH o placebo (doble enmascaramiento o ciego). c)Asignación aleatoria del NFH/placebo, lo que garantiza que los participantes de los distintos grupos de tratamiento son comparables en sus factores de riesgo cardiovascular, sean conocidos o no.
8. Seguimiento y exposiciónLos CVOT son estudios de larga duración en los que es esencial evaluar la exposición al NFH, es decir, conocer cuántos participantes y por cuánto tiempo lo recibieron (tabla 2). El CVOT que incluyó más participantes fue el DECLARE22, y el que menos, el SUSTAIN-617. El de mayor seguimiento mediano fue el REWIND23, y el de menor, Harmony24. El de menor porcentaje de abandonos de tratamiento fue el Saxagliptin Assessment of Vascular Outcomes Recorded in patients with diabetes mellitus-TIMI 53 (SAVOR)29, y el de mayor, el Exenatide Study of cardiovascular Events Lowering (EXSCEL)25. La mejor forma de notificar la exposición es proporcionarla en participantes-año, aunque a menudo es un dato que no se reporta o es difícil de encontrar en las publicaciones. Una baja exposición al NFH debe tenerse en cuenta al valorar los efectos sobre el OP. También debe evaluarse el porcentaje de participantes aleatorizados con información sobre su estado vital al final del ECA, aunque ha sido muy elevado en todos los CVOT.
9. Objetivo primarioTodos los CVOT han tenido como OP el tiempo hasta la aparición de uno de los eventos que forman ese OP, habitualmente MACE-3, salvo en el Trial Evaluating Cardiovascular Outcomes with Sitagliptin (TECOS), que añadió la hospitalización por angina inestable al MACE-3 en su OP28, y el DECLARE, que al final evaluó un objetivo coprimario de MACE-3 y un compuesto de MCV o HIC22. Para cada participante: a)Se considera tanto si aparece el evento como cuándo lo hace. b)Solo se considera el primer componente del OP que aparece, si es que aparece. Por ejemplo, si primero presenta un IM y luego un ACV o una MCV, solo se considerará que ha sufrido un IM. La comparación entre grupos siempre se hace con modelos de regresión de Cox (o de riesgo proporcionales), que aportan los resultados como HR (o cociente de riesgo instantáneo). Aunque la HR se asimila al RR, no son exactamente iguales, pues para calcular la HR se tiene en cuenta el momento en que se produce el evento, mientras que el RR se calcula en un determinado momento del seguimiento (p.ej., a los 2años) considerando el cociente de la proporción de eventos entre los dos grupos en ese momento pero sin considerar cuándo se produjeron los eventos38. Al evaluar la HR hay que considerar también el límite superior de su IC95%, pues es una medida de su significación estadística (es decir, su valor p). Ese límite fue de 0,99 en dos de los CVOT, indicando un resultado en el límite de la significación estadística (tabla 2)15,23.
Otra manera, menos común, de reportar el efecto cardiovascular beneficioso de un NFH es proporcionar el número de participantes que es necesario tratar para prevenir un evento del OP (number needed-to-treat). Este dato es de alto interés clínico. No puede calcularse fácilmente como la inversa de la reducción en el RA que consigue el NFH39, y debe ser proporcionado por los autores. Además, solo debería calcularse para el OP del CVOT. No debería calcularse para los objetivos secundarios. Y tampoco para resultados de los análisis de subgrupos, como se ha hecho recientemente en uno de los CVOT analizados23.
Todos los CVOT tienen OP compuestos. Esto permite detectar mayor número de eventos en menos tiempo, aumentado la potencia del estudio. Además, se puede capturar mejor el efecto del NFH, aunque los componentes más leves (p.ej., hospitalización por angina inestable en el TECOS)28 tienden a aparecer antes en el tiempo, diluyendo el efecto del NFH y favoreciendo la no inferioridad32. En todo caso, se deben mostrar los resultados de cada uno de los componentes del OP, para ver si todos ellos tienen el mismo sentido. El único CVOT en el que eso no ocurrió fue en el EMPA-REG, donde con su NFH se registró un aumento no significativo de ACV no fatal con descenso no significativo de IM no fatal y significativo de MCV, lo que restaría solidez a su resultado15.
10. Análisis de sensibilidad y análisis de subgruposPara aumentar la robustez del resultado sobre el OP es aconsejable realizar análisis de sensibilidad que lo confirmen40. Esto es, hacer análisis en los que solo se considere la población por protocolo, o por intención de tratar, o evaluar los datos en función de los participantes perdidos. Si estos análisis arrojan el mismo resultado que el inicial, se reforzaría este. En el EMPA-REG los resultados por protocolo no fueron significativos, pero sí lo fueron por intención de tratar15.
También conviene realizar análisis de subgrupos32. Estos análisis exploran la posible heterogeneidad del efecto del NFH sobre el OP en diferentes subgrupos de participantes definidos por sus características basales, tales como intervalo de edad, sexo, origen geográfico o ausencia de eventos cardiovasculares previos (prevención primaria). Este análisis se realiza mediante el cálculo de una p de interacción: si es significativa indicaría que los participantes se comportarían de forma diferente en función de la característica que define al subgrupo. Esto ocurrió, por ejemplo, en el EMPA-REG al analizar el OP en función del origen geográfico de los participantes15. Este tipo de análisis es solo exploratorio y plantea el problema de las comparaciones múltiples. Cuando se hacen 10 análisis de este tipo en un mismo estudio considerando como significativa una p de interacción <0,05, el riesgo de obtener al menos un falso positivo es >40%41. El manejo de este problema se desarrolla en el punto12. Al ser exploratorio, si se confirma, por ejemplo, que el subgrupo de participantes en prevención primaria se comporta igual que en prevención secundaria, este hecho no permite afirmar que un efecto beneficioso sobre el OP detectado al analizar ambos subgrupos a la vez sea extrapolable al subgrupo en prevención primaria (donde siempre es más difícil demostrar cualquier beneficio cardiovascular). Esto ocurrió en el REWIND, el CVOT con mayor porcentaje de participantes en prevención primaria de todos los realizados, donde se comentó que su NFH mejoraba significativamente el OP en el subgrupo en prevención primaria del estudio23. Un metaanálisis posterior ha analizado los efectos cardiovasculares de todos los CVOT con ARGLP1 según los participantes estuvieran en prevención primaria (la mayoría pertenecían al REWIND) o secundaria, concluyendo la ausencia de beneficio en prevención primaria con beneficio en prevención secundaria42.
11. Diseños flexibles (adaptive designs)Los diseños flexibles pueden mejorar la eficiencia de los ECA, con potenciales beneficios para los participantes, reduciendo costes y aumentando las posibilidades de encontrar posibles beneficios, en caso de existir. Los hay de diverso tipo, pero siempre deberían preespecificarse o, si el ECA ya está en curso, realizarse sin conocer sus resultados43. Este tipo de diseño se realizó en el EMPA-REG, cuando se retiró del OP los infartos silentes antes de publicar los resultados. Los infartos silentes fueron más frecuentes en los grupos con NFH que en el que recibió placebo (7,0 frente a 5,4/1.000 participantes-año), pero no podemos saber si el no haber hecho ese cambio habría o no modificado el resultado final del estudio, que estuvo en el límite de la significación estadística, pues no se ha reportado cuánto tiempo tardaron en aparecer15. Otro ejemplo es el del DECLARE, que inicialmente se planteó con el OP de no inferioridad para MACE-322. Durante el ensayo, a raíz de los resultados del EMPA-REG sobre HIC, y sin conocer sus resultados, se añadió un objetivo compuesto de MCV o HIC como coprimario del MACE-3 inicial. Para ajustar el error alfa se bajó a la mitad su valor inicial de 0,05, de forma que se iba a exigir al final del ensayo a ambos objetivos coprimarios un valor de p<0,0231 para considerarlos significativos, siendo necesario que lo fueran ambos para analizar jerárquicamente el efecto sobre una serie de objetivos secundarios ya preespecificados al inicio del ensayo (con p<0,05)44. En este ensayo, MACE-3 no fue significativo (p=0,17), pero sí lo fue el otro coprimario de MCV o HIC (p=0,005).
12. Objetivos secundarios y exploratoriosTodos los CVOT incluyen objetivos secundarios. Todos ellos son exploratorios y generadores de hipótesis, aunque si su análisis arrojara un valor de p muy bajo (p.ej., <0,001) habría que considerar que podrían ser significativos. Tanto en el EMPA-REG15 como en el CANVAS16 la HIC fue un objetivo secundario en el que se observó un notable beneficio del respectivo NFH, y que ha dado pie a ensayos específicos para el tratamiento de la insuficiencia cardíaca45.
Como objetivos secundarios se recomienda incluir todos los componentes del OP. Para que agencias como la FDA consideren que sus resultados son suficientes como para modificar la ficha técnica (p.ej., como ocurrió con la MCV y el EMPA-REG), pueden seguirse dos estrategias32: a)Preespecificar una jerarquía de evaluación de objetivos secundarios sin modificar el valor de p inicial, pero de forma que, para pasar de un objetivo al siguiente, el primero de ellos deba ser significativo; esta es la estrategia más habitual en los CVOT. b)No realizar esa jerarquía, pero ajustar el valor de p a la baja en función del número de objetivos secundarios (comparaciones múltiples) que se preespecifiquen. Por ejemplo, si el valor inicial de p es <0,05 y existen cinco objetivos secundarios, para aceptar cualquiera de ellos como significativo su valor p debería ser <0,01. Probablemente, esta estrategia sería la más neutra44.
13. Evaluación de eventos adversosEn los CVOT pueden confirmarse efectos adversos ya conocidos de los NFH que se evalúan, como las infecciones genitales de los ISGLT2, o la intolerancia digestiva de los ARGLP1. Además, al incluir miles de participantes pueden también ayudar a detectar potenciales nuevos efectos adversos, se confirmen luego o no, como desarrollo de retinopatía17 o aumento de amputaciones16. También hay que estudiar su aparición en el grupo con placebo, pues si por ejemplo tiene un aumento de hipoglucemias con relación al grupo con el NFH, esto puede ayudar a explicar parte de los resultados26.
La nueva propuesta de la FDATras una reunión de un comité de expertos en octubre de 2018, que constató que ninguno de los CVOT hechos con la guía de 2008 detectó un aumento de riesgo cardiovascular con los NFH, así como la necesidad de evaluar personas con DM2 con mayor riesgo de efectos adversos y otros tipos de efectos adversos (y no solo cardiovasculares), en marzo de 2020 la FDA emitió unas nuevas recomendaciones sobre cómo evaluar la seguridad de los NFH, que aparecen resumidas en la tabla 346. Estas nuevas recomendaciones sustituyen a las de 2008. En ellas no se proponen puntos de corte para establecer la seguridad del NFH y se indica que la valoración de la seguridad será flexible. Con estas recomendaciones se establece como fulcro una exposición mínima de participantes al NFH de 4.000 pacientes-año, que en general sería más baja que la registrada en los CVOT comentados previamente, pero se asegura que participen un número mínimo de personas con insuficiencia renal, enfermedad cardiovascular o edad superior a 65años. Con esas recomendaciones, los nuevos ensayos clínicos podrían precisar evaluar menos eventos cardiovasculares para poder conseguir la autorización de sus respectivos NFH47. Ha sorprendido que se hayan abandonado los puntos de corte de HR para el OP, así como que no se haya recomendado realizar ensayos pragmáticos (ensayos que preservan la aleatorización pero se imbrican en la práctica clínica habitual) como estrategia para incluir participantes lo más parecidos posible a los potenciales candidatos a recibir el NFH48.
Propuesta de la FDA de marzo de 2020 para el estudio de la seguridad de los NFH, que sustituye a la previa de 2008 para el estudio de la seguridad cardiovascular de los NFH46
| Recomendaciones de la FDA de marzo de 2020 para evaluar la seguridad de los nuevos fármacos hipoglucemiantes (NFH) |
|---|
| 1. Antes de su autorización para comercialización, los NFH deberían presentar ensayos clínicos controlados (extensiones incluidas) con al menos 4.000 pacientes-año expuestos al NFH en ensayos en fase3 (todas las dosis son válidas), con por lo menos 1.500 pacientes expuestos al menos 1año y por lo menos 500 pacientes expuestos 2años |
| 2. Inclusión de pacientes con características especiales:• Al menos 500 con insuficiencia renal crónica estadios 3 o 4• Al menos 600 con enfermedad cardiovascular establecida (infarto de miocardio, accidente cerebrovascular o enfermedad arterial periférica previas, o bien enfermedad coronaria documentada)• Y al menos 600 pacientes mayores de 65años• Al menos 1.200 pacientes deben poder enmarcarse en una de esas tres categorías |
| 3. La evaluación de los efectos adversos cardiovasculares sigue siendo importante. Si es esperable un efecto adverso peculiar del NFH, se recomienda contactar con la FDA para el diseño de los ensayos en fase3 |
| 4. La evaluación de los posibles efectos adversos del NFH sigue precisando del concurso de comités independientes |
En resumen, para valorar mejor el potencial impacto en la práctica clínica de los CVOTs hay que considerar que:
- 1.
Todos los análisis deben preespecificarse; los análisis post-hoc siempre son exploratorios.
- 2.
Demostrar no inferioridad requiere mayor tamaño muestral que demostrar superioridad.
- 3.
Es más importante conocer la tasa de eventos en el grupo control (placebo) que el porcentaje de participantes en prevención primaria, como también es importante valorar la exposición al NFH.
- 4.
Los períodos de run-in restan validez externa a los resultados del estudio.
- 5.
En los CVOT siempre se intenta minimizar las diferencias en HbA1c entre grupos, y no son aptos para medir la eficacia de los NFH sobre la glucemia.
- 6.
Todos ellos finalizan cuando se alcanza un número de eventos previamente especificado.
- 7.
El OP siempre tiene varios componentes, y en cada participante solo se tiene en cuenta el primero de los componentes que se produce y cuándo se produce. Al valorar la HR del OP, si el límite superior de su IC95% está próximo a 1,0 (p.ej., 0,99) indica que el resultado estuvo en el límite de la significación estadística. Clínicamente, es muy interesante conocer el número de participantes que se debería tratar para prevenir un componente del OP.
- 8.
Los análisis de sensibilidad pueden dar robustez a los resultados obtenidos en el OP; los de subgrupos en relación al OP y los de los objetivos secundarios son exploratorios y generadores de hipótesis.
- 9.
Es importante comprobar si se produjeron cambios en el OP a lo largo del ensayo. Estas consideraciones pueden ayudar a evaluar de forma más objetiva los CVOT, tarea aún propia y exclusiva del pensamiento crítico humano, y no asumible por la inteligencia artificial49.
El presente trabajo ha sido financiado por el PI15/00567 (IP: JMCG) del Fondo de Investigación Sanitaria (FIS), a su vez cofinanciado por el Instituto de Salud CarlosIII-Rama de Evaluación General (Ministerio de Economía y Competitividad) y el Fondo Europeo de Desarrollo Regional (FEDR).
Conflicto de interesesAR: Ninguno. GL: ha recibido financiación por ponencias o asistencia a congresos/cursos médicos desde 2018 de Abbott, Janssen y Sanofi. JMGC ha recibido financiación por ponencias, asistencia a comités de expertos o a congresos/cursos médicos desde 2018 de AstraZeneca, Janssen, Menarini, MSD, Novo Nordisk y Sanofi. JMGC ha participado como investigador en el CANVAS program.
