




Maturity-onset diabetes of the young (MODY) comprises a heterogeneous group of monogenic disorders characterized by primary defect in pancreatic β-cell function, early onset and autosomal dominant inheritance, accounting for about 1–5% of all diabetes diagnoses. Mutations in 14 genes are responsible for the majority of all MODY cases described so far. The clinical phenotype relies on genetic defects, with important implications in the optimal treatment and prognosis definition. MODY's early diagnosis remains a challenge, since this group of inherited disorders comprises a large clinical spectrum and it usually overlaps with other types of diabetes, requiring a high index of suspicion even if the definitive statement demands a molecular genetic study.
Recent advances on the genetic determinants and pathophysiology of MODY have allowed a better understanding of its underlying molecular mechanisms, providing a proper genetic counseling and early diagnosis. These new management insights will make possible to set up new therapeutic strategies, with drugs able to prevent, correct or at least delay the decline of pancreatic β-cell function, thus affording for a more personalized treatment and, ultimately, for a better patient care.
La diabetes tipo MODY (del inglés, maturity onset diabetes of the young) comprende un grupo heterogéneo de enfermedades monogénicas caracterizadas por un defecto primario de la función de las células β-pancreáticas, con inicio precoz y herencia autosómica dominante, que representa aproximadamente el 1-5% de las diabetes diagnosticadas. Las mutaciones descritas hasta la actualidad en 14 genes son responsables de la mayoría de los casos de la diabetes tipo MODY. El fenotipo clínico depende del defecto genético, y su identificación es importante para definir el tratamiento y el pronóstico. El diagnóstico temprano de la diabetes tipo MODY sigue siendo un reto, puesto que abarca un largo espectro de manifestaciones que se solapan con las de otros tipos de diabetes, exigiendo así un elevado grado de sospecha clínica y el diagnóstico definitivo necesita del estudio genético molecular.
Los progresos recientes en el conocimiento de los determinantes genéticos y de la fisiopatología de la diabetes tipo MODY han permitido una mejor comprensión de los mecanismos moleculares subyacentes de estos procesos, y han facilitado un adecuado asesoramiento genético y el diagnóstico precoz. Estos nuevos conocimientos el abordaje permitirán el desarrollo de nuevas estrategias terapéuticas con fármacos capaces de prevenir, reparar o al menos retrasar el deterioro de la función de las células β-pancreáticas. Todo ello posibilitará un tratamiento más personalizado y, en última instancia, una mejor atención del paciente.
Maturity-onset diabetes of the young (MODY) is a monogenic form of diabetes that comprises a heterogeneous group of disorders characterized by primary defect in pancreatic β-cell function, early onset (classically presenting before the age of 25) and autosomal dominant inheritance, with absence of autoimmunity or, for most of the cases, ketosis.1,2 Other monogenic forms of diabetes comprises neonatal diabetes, syndromic forms and mitochondrial diabetes (maternally inherited diabetes and deafness).3 Commonly misdiagnosed as Type 1 Diabetes Mellitus (T1DM) or Type 2 Diabetes Mellitus (T2DM), MODY has an estimated prevalence of 1–5% in the diabetic population, thought frequently undervalued.4,5
The molecular basis of MODY was recognized a couple decades ago, since genetic mutations result in diabetes primarily through their effects on β-cell dysfunction. Moreover, the clinical features of patients with MODY are currently known to be heterogeneous, depending on the genetic etiology.6,7
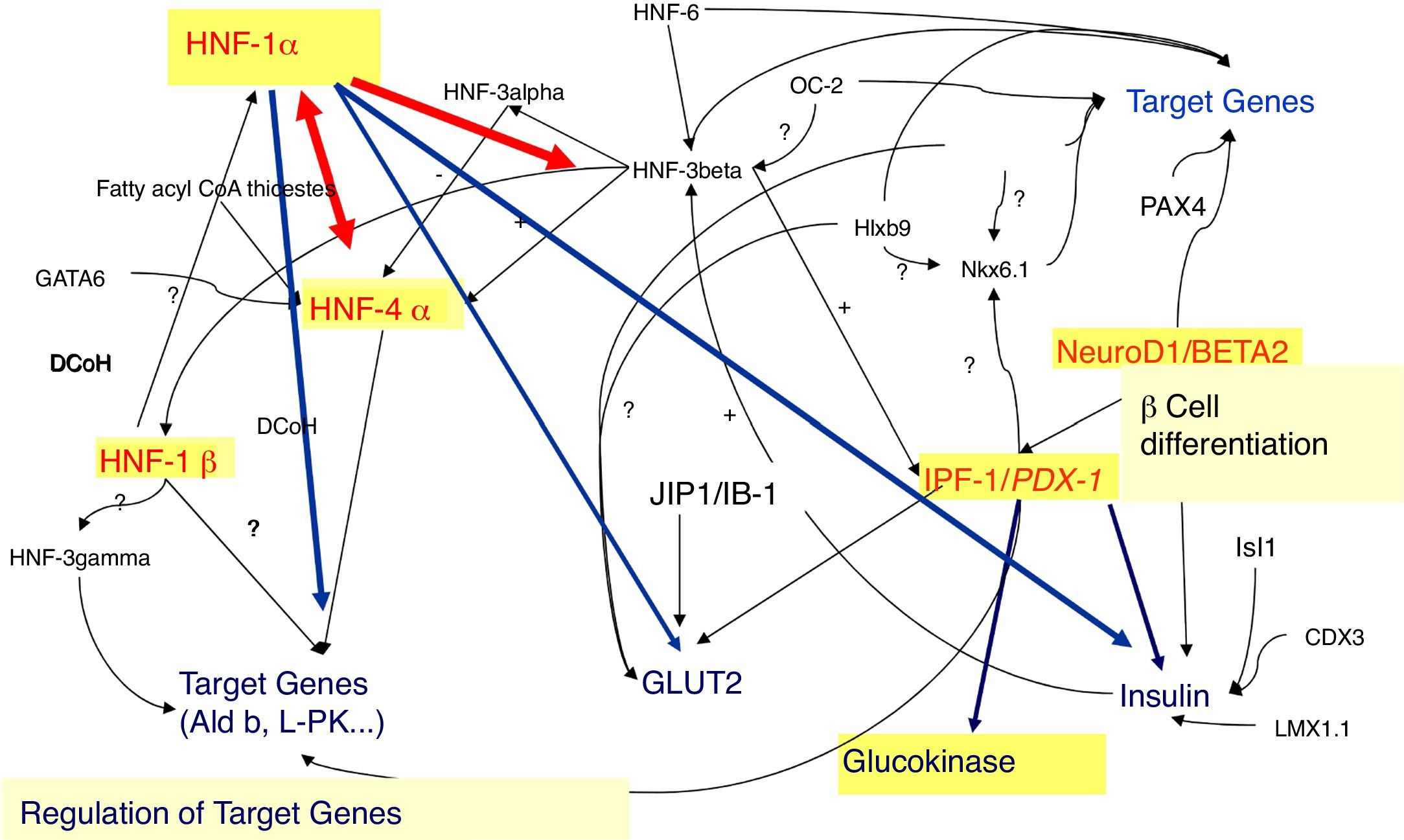
In fact, 14 MODY subtypes has been described so far, referred from 1 to 14, according to the order of the discovery, with mutations in 14 different genes (Table 1, Fig. 1). Several of these genes encodes for transcription factors, namely: hepatocyte nuclear factor (HNF) 4α (related to MODY 1), HNF-1α (related to MODY 3), insulin promoter factor 1 (IPF-1) (related to MODY 4), HNF-1β (related to MODY 5), neurogenic differentiation factor 1 (NeuroD1), also known as β-cell E-box transactivator 2 (BETA2) (related to MODY 6), Kruppel-like factor 11 (KLF11) (related to MODY 7) and paired box gene 4 (PAX 4) (related to MODY 9). With regard to the others, glucokinase (GCK) gene encodes GCK enzyme (related to MODY 2). Insulin (INS) gene encodes for proinsulin precursor of insulin (related to MODY 8). Carboxyl-ester lipase (CEL) gene encodes CEL lipase enzyme (related to MODY 10). B lymphoid tyrosine kinase (BLK) gene encodes BLK protein (related to MODY 11). ATP-binding cassette transporter sub-family C member 8 (ABCC8) gene encodes ABCC8 protein (related to MODY 12). Potassium voltage-gated channel subfamily J member 11 (KCNJ11) gene encodes human BIR (β-cell inward rectifier or Kir6.20) (Fig. 2) (related to MODY 13). Finally, Adaptor Protein Phosphotyrosine interacting with PH domain and Leucine zipper 1 (APPL1) gene (related to MODY 14) encodes APPL1 adapter protein.4,8
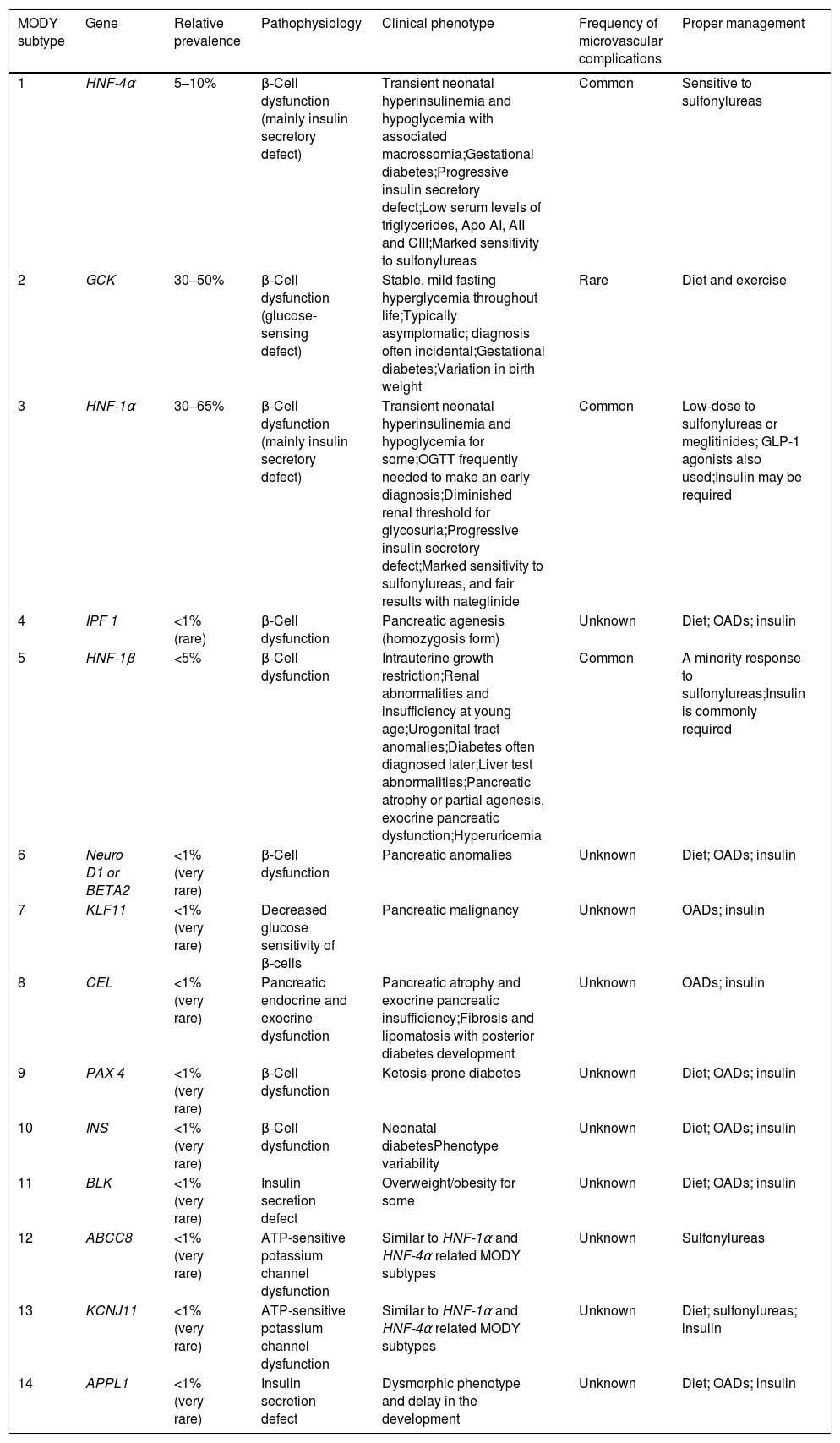
The different MODY subtypes and their related molecular basis, clinical features and proper management.
| MODY subtype | Gene | Relative prevalence | Pathophysiology | Clinical phenotype | Frequency of microvascular complications | Proper management |
|---|---|---|---|---|---|---|
| 1 | HNF-4α | 5–10% | β-Cell dysfunction (mainly insulin secretory defect) | Transient neonatal hyperinsulinemia and hypoglycemia with associated macrossomia;Gestational diabetes;Progressive insulin secretory defect;Low serum levels of triglycerides, Apo AI, AII and CIII;Marked sensitivity to sulfonylureas | Common | Sensitive to sulfonylureas |
| 2 | GCK | 30–50% | β-Cell dysfunction (glucose-sensing defect) | Stable, mild fasting hyperglycemia throughout life;Typically asymptomatic; diagnosis often incidental;Gestational diabetes;Variation in birth weight | Rare | Diet and exercise |
| 3 | HNF-1α | 30–65% | β-Cell dysfunction (mainly insulin secretory defect) | Transient neonatal hyperinsulinemia and hypoglycemia for some;OGTT frequently needed to make an early diagnosis;Diminished renal threshold for glycosuria;Progressive insulin secretory defect;Marked sensitivity to sulfonylureas, and fair results with nateglinide | Common | Low-dose to sulfonylureas or meglitinides; GLP-1 agonists also used;Insulin may be required |
| 4 | IPF 1 | <1% (rare) | β-Cell dysfunction | Pancreatic agenesis (homozygosis form) | Unknown | Diet; OADs; insulin |
| 5 | HNF-1β | <5% | β-Cell dysfunction | Intrauterine growth restriction;Renal abnormalities and insufficiency at young age;Urogenital tract anomalies;Diabetes often diagnosed later;Liver test abnormalities;Pancreatic atrophy or partial agenesis, exocrine pancreatic dysfunction;Hyperuricemia | Common | A minority response to sulfonylureas;Insulin is commonly required |
| 6 | Neuro D1 or BETA2 | <1% (very rare) | β-Cell dysfunction | Pancreatic anomalies | Unknown | Diet; OADs; insulin |
| 7 | KLF11 | <1% (very rare) | Decreased glucose sensitivity of β-cells | Pancreatic malignancy | Unknown | OADs; insulin |
| 8 | CEL | <1% (very rare) | Pancreatic endocrine and exocrine dysfunction | Pancreatic atrophy and exocrine pancreatic insufficiency;Fibrosis and lipomatosis with posterior diabetes development | Unknown | OADs; insulin |
| 9 | PAX 4 | <1% (very rare) | β-Cell dysfunction | Ketosis-prone diabetes | Unknown | Diet; OADs; insulin |
| 10 | INS | <1% (very rare) | β-Cell dysfunction | Neonatal diabetesPhenotype variability | Unknown | Diet; OADs; insulin |
| 11 | BLK | <1% (very rare) | Insulin secretion defect | Overweight/obesity for some | Unknown | Diet; OADs; insulin |
| 12 | ABCC8 | <1% (very rare) | ATP-sensitive potassium channel dysfunction | Similar to HNF-1α and HNF-4α related MODY subtypes | Unknown | Sulfonylureas |
| 13 | KCNJ11 | <1% (very rare) | ATP-sensitive potassium channel dysfunction | Similar to HNF-1α and HNF-4α related MODY subtypes | Unknown | Diet; sulfonylureas; insulin |
| 14 | APPL1 | <1% (very rare) | Insulin secretion defect | Dysmorphic phenotype and delay in the development | Unknown | Diet; OADs; insulin |
MODY, maturity-onset diabetes of the young; OADs, oral antidiabetic drugs; OGTT, oral glucose tolerance test; Apo AI, AII and CIII, apolipoproteins AI, AII and CIII; Lp(a), lipoprotein Lp(a); TG, triglycerides; HNF, hepatocyte nuclear factor; GCK, Glucokinase; IPF-1, insulin promoter factor-1; NeuroD1, neurogenic differentiation factor 1; BETA2, β-cell E-box transactivator 2; KLF11, Kruppel-like factor 11; CEL, bile salt dependent lipase; PAX 4, paired box gene 4; INS, insulin; BLK, B lymphoid tyrosine kinase gene; ABCC8, ATP-binding cassette transporter sub-family C member 8 gene; KCNJ11, potassium voltage-gated channel subfamily J member 11 gene; APPL1, Adaptor Protein, Phosphotyrosine interacting with PH domain and Leucine zipper 1.

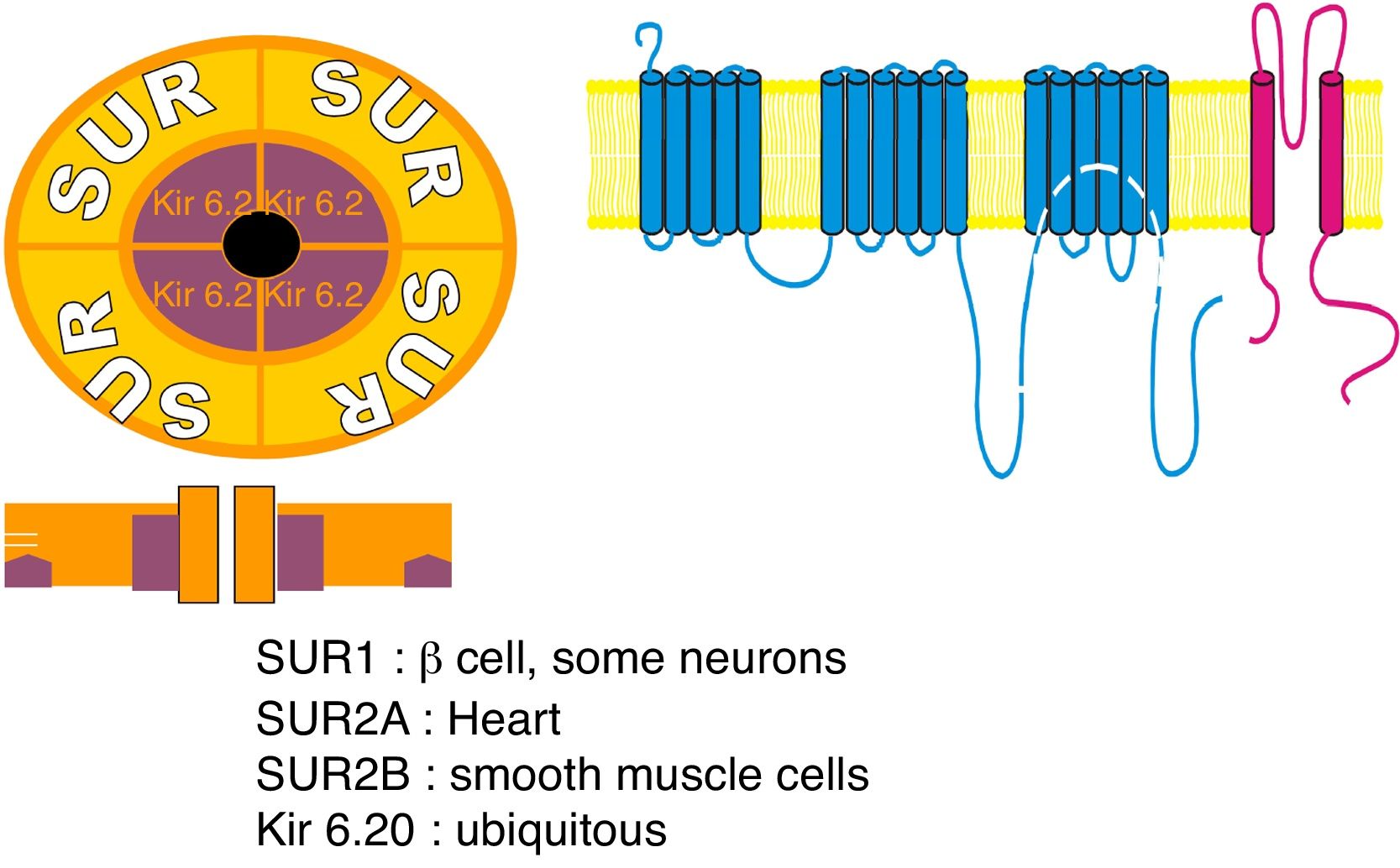
 components: 1 – SUR encoded by ABCC8 gene; 2 – potassium voltage-gated channel; Kir 6.20 – encoded by subfamily J member 11 (KCNJ11) gene.")
Sulfonylurea receptor (SUR) components: 1 – SUR encoded by ABCC8 gene; 2 – potassium voltage-gated channel; Kir 6.20 – encoded by subfamily J member 11 (KCNJ11) gene.
Among these, the most common MODY subtypes are MODY 3 and MODY 2. MODY 1, 5, and 4 are rare, whereas MODY 6–14 comprises a group of very rare forms of MODY.5,8
MODY's early diagnosis still remains a challenge for physicians, with important future implications, since it will allow treatment optimization, prognosis definition and genetic counseling of family members.1,7
In this sense, this review intends to be an update of the state of art of all currently known MODY's subtypes and the way of dealing with, also seeking to disclose MODY's future management based on the latest research on this field.
Gene mutations involved in the different MODY's forms, the respective phenotype outcomes and managementHNF-1α and GCK products represents the most common forms of MODY phenotypes (30–65% and 30–50%, respectively), followed by HNF-4α (5–10%) and HNF-1β (<5%).8 The remaining genetic subtypes, being rarer causes of MODY (<1%), have limited data available with few published literature.8
HNF-1α and HNF-4α (MODY 3 and MODY 1)The hepatic transcription factors HNF-1α and HNF-4α are responsible for the gene's regulation in the liver, pancreatic islet cells, kidneys, and genital tissues.9–11 Mutations in the genes encoding these transcription factors may account for some MODY subtypes, such as MODY 3 and 1, respectively.9–11
In fact, in pancreatic β-cells, these transcription factors regulate the expression of the INS gene, as well as the expression of genes encoding proteins involved in the transport and metabolism of glucose.9,10,12 In the liver, these proteins regulate lipoprotein biosynthesis.9,10 The expression of HNF-1α is partially regulated by HNF-4α.11,13
The pathophysiological mechanisms of MODY related with mutations in the HNF-1α (MODY 3) and HNF-4α (MODY 1) genes are very similar, since HNF-1α expression is regulated by HNF-4α.11,13
Fasting hyperglycemia is relatively mild in these patients, albeit they have significantly higher glycemic levels 2h after oral glucose overload compared to those with mutated GCK.12,14 In MODY 3 and MODY 1 hyperglycemia tends to increase over time and, thus, for most patients, pharmacological treatment with oral antidiabetic drugs (OADs) or insulin will be required (30–40% demands insulin).14,15 Evidence has shown that glucose-induced insulin secretion declines by 1–4% per year in patients with MODY 3.15,16 In addition, along with transient neonatal hyperinsulinemic hypoglycemia, macrosomia, and gestational diabetes, are also common features of these MODY subtypes.14,17
These patients may have the full spectrum of diabetes complications. In fact, microvascular complications, particularly retinopathy and nephropathy, are as frequent as in T1DM or T2DM14,16 (Table 1).
In addition to the effects in β-cells, since HNF-1α and HNF-4α are also expressed in other organs, these subjects present extrapancreatic manifestations, affecting renal and hepatic functions.12,18 In fact, reduced glucose renal reabsorption (i.e., a low renal threshold for glucose) and glycosuria represents a MODY 3 hallmark.12,19 On the other hand, HNF-4α deficiency is related with a reduction in triglyceride concentration, as well as in serum levels of apolipoproteins AI, AII, CIII, and lipoprotein Lp(a)20 (Table 1).
In regard to treatment, an important distinctive feature of HNF-1α mutations is the particularly sensitivity to the hypoglycemic effects of sulfonylureas.12,21 In a randomized cross-over study comparing individuals diagnosed with MODY 3 with those with T2DM, a 4-fold increased response to gliclazide was found for MODY 3 subjects, whereas the response to metformin was similar in both groups.21Nateglinide has also been used in these patients, suggesting that prandial secretagogues may be a useful alternative.16,22 Although glycemic control could be sustained for several years, most patients will eventually move toward insulin treatment due to a progressive β-cell dysfunction.16
Similarly, a low dose of sulfonylureas (12.5% or less of the maximum authorized dose) seems to be an effective treatment for patients with HNF-4α mutations.14
GCK (MODY 2)GCK enzyme is expressed, at high concentrations, in pancreatic and hepatic cells.18 It catalyses the phosphate transfer from ATP to glucose, generating glucose-6-phosphate, whose metabolism, in turn, stimulates the secretion of insulin by β-cells.18,23 Therefore, GCK acts as a “glucose sensor” in β-cells. Moreover, in the liver, GCK plays a key role in the glucose hepatic storage as glycogen, particularly in the postprandial state.16,18 Heterozygous inactivating mutations in the GCK gene leads to a partial deficiency of this enzyme, which is responsible for MODY 2; homozygous inactivating mutations induce the complete GCK deficiency and consequently, neonatal diabetes mellitus.4,23
GCK-related MODY (MODY 2) is a common form, especially in children and women with history of gestational diabetes.12,16 In MODY 2, hyperglycemia is mild, non-progressive, and asymptomatic at the time of diagnosis, not associated with vascular complications common in other types of diabetes.7,24 Less than 50% of genetic carriers will develop diabetes, whereas nearly 50% of women may develop gestational diabetes.18 Noteworthy, in MODY 2 families, birth weight of newborn infants relies on both fetal and maternal mutation status. In the absence of GCK mutation in the baby, he may be at risk for macrosomia and increased birth weight in approximately 500g, as a result of the exposure to maternal hyperglycemia. When the mutation is inherited from the mother, baby has similar homeostatic glucose set points and, thus, normal birth weight. On the other hand, if the mutation is inherited from the father, insulin secretion in the baby is reduced and birth weight is decreased in about 500g16 (Table 1).
The general consensus has pointed out that for most patients with MODY 2 pharmacological treatment will not be necessary.12,14 Exceptionally, some subjects will demand insulin therapy, namely in cases of severe hyperglycemia (≤2% of MODY 2) and pregnant women, in which insulin treatment could be necessary in order to prevent excessive fetal growth.12,18
HNF-1β (MODY 5)HNF-1β gene belongs to the homeodomain-containing family of transcription factors, being involved in embryonic development of several organs, including kidneys, urinary tract, liver, and pancreas.25 Mutations in this gene may account for another MODY subtype, designated MODY 5.18,26 Its plausible that the affected subjects may develop renal disease, characterized by renal cysts, renal dysplasia, renal malformations, or hypoplastic glomerulocystic kidney disease. In fact, HNF-1β mutation is specifically associated with non-diabetic renal disease, with renal function ranging from mild to terminal renal insufficiency.16,27 Pancreatic atrophy, abnormalities of the female genital tract and abnormal liver function among affected individuals can also be observed.14,25 Birth weight is reduced by nearly 900g due to a decreased insulin secretion in uterus.4,7 About a half of MODY 5 subjects present diabetes at youth, as those with MODY 3.14 However, unlike the latter, HNF-1β mutations are related with insulin resistance and hyperuricemia.16,28
Thereby, the low insulin sensitivity of MODY 5 patients suggests an insulin sensitizer, such as metformin or pioglitazone, as the oral drug of choice.28 Actually, these subjects do not respond particularly well to sulfonylureas, and usually require early insulin therapy.7,24
IPF-1 (MODY 4)IPF-1 is a pancreatic homeodomain transcription factor (also known as IDX-1, STF-1 and PDX-1) that regulates both early pancreatic development and the expression of key endocrine β-cell-specific genes, most notably insulin.29 Targeted disruption of the IPF-1 gene in mice results in pancreatic agenesis. In a human subject with pancreatic agenesis, this phenotype was attributable to homozygosity for an inactivating mutation of IPF-1 gene. Heterozygous carriers of the mutation develop MODY 4.29
The current insights of IPF-1-related MODY (MODY 4) are mainly based on studies in a few families, including a family with MODY 2 and MODY 4 combined mutations.29–32 Molecular studies in a child with neonatal diabetes and exocrine pancreatic insufficiency – as a result of congenital agenesis of the pancreas – revealed the presence of the homozygous form of the IPF-1 mutation; the parents were carriers of the heterozygous form of this mutation.33 The whole family evaluation had revealed a high prevalence of a mild form of diabetes, with autosomal dominant transmission, along with the heterozygous mutation in the IPF-1.33 For the carriers of this mutation, the onset of diabetes may occur at more advanced ages (around the age of 35), compared to other MODY subtypes, and can be treated with diet, OAD and/or possibly insulin, for the most severe cases.7,33 In an Italian family with Pro to Thr substitution (P33T) in the IPF1 transactivation domain the clinical phenotype goes from gestational diabetes, MODY4 to T2DM.31
NeuroD1 (MODY 6)The transcription factor NeuroD1 (also known as BETA2) was isolated based on its ability to activate the transcription of the INS gene, being fundamental for the normal development of pancreatic islets.34 Mutations in NeuroD1 encoding genes may be responsible for another MODY subtype, designated MODY 6.18
NeuroD1-related MODY is characterized by permanent neonatal diabetes and a consistent pattern of neurological abnormalities including cerebellar hypoplasia, learning difficulties, sensorineural deafness, and visual impairment.16,35,36 This syndrome highlights the key role of NeuroD1 in both the development of the endocrine pancreas and the central nervous system in humans.36 The optimal therapeutic approach is yet to be disclosed, since the related literature is scarce though it suggests OAD or insulin requirement.7,35
KLF11 (MODY 7)KLF11 is a transcription factor present in pancreatic β-cells that acts as a negative regulator of exocrine cell growth.37 KLF11 also plays a key role as a glucose-induced regulator of INS gene, through binding to insulin promoter, as other MODY subtypes already mentioned.38 In addition, increased repression of the catalase 1 promoter has been reported, suggesting a role in the clearance of free radicals that can turn these cells more susceptible to oxidative stress.4,37KLF11-related MODY is often associated with pancreatic malignancy.37
CEL (MODY 8)CEL gene encodes CEL enzyme involved in digesting milk and hydrolyzing dietary esters in duodenum and is also responsible for the hydration and absorption of cholesterol and liposoluble molecules.39 Current literature supports the involvement of CEL, also known as bile salt-dependent lipase (BSDL), in the pathophysiology of these pancreatic diseases.40 This enzyme is normally secreted by the exocrine pancreas and is diverted within the intestinal lumen to participate in the hydrolysis of dietary lipids. CEL is not transcribed into β-cells, being mainly expressed in pancreatic acinar tissues and lactating mammary glands.39 Despite the underlying pathophysiological mechanisms are yet to be fully understood, mutations in this gene are responsible for an exocrine and endocrine pancreatic dysfunction associated with another MODY subtype, MODY 8.4,39,41
CEL-related MODY is associated with pancreatic atrophy, fibrosis and lipomatosis together with exocrine insufficiency and later endocrine dysfunction and diabetes. Fat infiltration is an early event in non-diabetic carriers of mutations in the variable number of tandem repeat (VNTR) region of CEL, with signs of exocrine dysfunction that meet the criteria for chronic pancreatitis.40,41 Dyslipidemia is associated with an inflammatory status in patients with diabetes. It is known that branched fatty esters of hydroxyl fatty acids (FAHFAs) present protective effects against diabetes, with anti-inflammatory activities.42 A recent study demonstrates that FAHFAs are the preferred substrates for CEL.42 Any perturbation in the BSDL variant homeostasis, associated with different enzymatic properties, leading to its retention within the cell, could induce active degradation of these protective endogenous FAHFAs. Suppression of the anti-inflammatory activity of FAHFAs upon BSDL hydrolysis may cause diabetes.40 Overall, these data suggested that deletion in VNTR, such as c.1686delT, leads to modification of the CEL specificity. Therefore, it can be hypothesized that any BSDL/CEL variant with VNTR mutations may have different specificity from that of the non-mutated enzyme.40
PAX4 (MODY 9)PAX 4 is a transcription factor member of PAX family that regulates fetal development, cancer growth and also represses the promoter activity of insulin and glucagon.43 PAX4 is required for the regeneration of β-cells in adults and its mutation blocks or inhibits β-cell growth and proliferation.43,44 In this sense, mutations in this gene leads to impaired glucose-dependent insulin secretion and were reported as the cause of a monogenetic form of diabetes, designated MODY 9.44,45 Some PAX4 mutations induced increased susceptibility of β-cell to apoptosis upon high glucose exposure. PAX4-related MODY 9 has been linked with ketosis-prone diabetes.46
INS (MODY 10)INS gene encodes the proinsulin precursor of insulin molecule.47,48 It has been reported that a mutation in this gene cause a defect in the nuclear factor kappa-light-chain-enhancer of activated β-cells (NF-κB) transcription factor, leading to reduced structural stability of insulin molecule associated with a very rare form of MODY subtype, designated as MODY 10.4,49INS-related MODY (MODY 10) is also linked with neonatal diabetes.48
BLK (MODY 11)BLK-B gene, expressed in pancreatic β-cells, encodes a nonreceptor tyrosine kinase of the src family of proto-oncogenes and plays a major role in thymopoiesis in immature T cells.4,50 Moreover, it promotes the glucose-dependent insulin synthesis and secretion through the upregulation of PDX-1 and NKx-6 transcription factors.50 The BLK gene mutation mainly affects MIN6 β-cells (a highly differentiated β-cell line) and, therefore, is responsible for another MODY subtype, MODY 11.50
BLK-related MODY is linked with a higher prevalence of the obese phenotype compared with other MODY's individuals.51
ABCC8 (MODY 12)ABCC8 gene encodes the sulfonylurea receptor 1 (SUR 1) subunit of ATP-sensitive potassium (KATP) channel found across pancreatic β-cells membrane, being involved in the insulin secretion process52,53 (Fig. 2). Mutations in this gene results in sulfonylureas responsive MODY, termed MODY 12, which clinical features are similar to those of HNF-1α/4α related MODY, being associated with neonatal diabetes, and can occur due to both activating or inactivating mutations of ABCC8 gene.4,52
KCNJ11 (MODY 13)KCNJ11 gene encodes Kir6.20 subunit of the pancreatic β-cells ATP-sensitive potassium (KATP) channel, playing a role in structure modulation of this channel and, thus, in insulin secretion54 (Fig. 2). Therefore, KCNJ11 gene mutations, responsible for another MODY subtype, designated MODY 13, leads to impaired fasting glucose or impaired glucose tolerance, due to the disrupted subunit interaction. Likewise INS-related MODY (MODY 10) and ABCC8-related MODY (MODY 12), MODY 13 is associated with neonatal diabetes being also sensitive to sulfonylurea therapy.4,54
APPL1 (MODY 14)APPL1 gene is widely expressed in all insulin target tissues and organs including the liver, adipose tissue, skeletal muscle, and pancreas.55 In pancreatic islets, APPL1 adapter protein co-localizes with insulin, indicating that is abundantly expressed in pancreatic β-cells where it acts as a physiological regulator of insulin secretion, binding to AKT2, a key molecule in the insulin signaling pathway. Mutations in APPL1 gene can cause APPL1 loss-of-function leading to MODY 14 phenotype.55 In addition, evidences from a zebrafish-based model have also linked APPL1 overexpression with dysmorphic phenotypes and delay in the development.56
MODY-xA significant percentage of patients with clinical characteristics of MODY, has an unknown causative gene (MODY-x).16,57 The identification of additional genes related to MODY will clarify the molecular basis of diabetes in these patients. Frayling et al. conducted a multicenter study with several researchers from Oxford, Lille and Malmo, stressing the need to continue the search for new mutations in families with MODY-x.58 In this study, 3 loci with Heterogeneity Logarithm of Odds (HLOD) scores >1.0 on chromosomes 3, 16 and 20 were identified, albeit the required level of significance of 3.3 has not been achieved. The identification of these 3 loci confirms the vast genetic heterogeneity in families with MODY-x.58
Further research will be needed in order to get a deeper knowledge on MODY's pathophysiological mechanisms and, therefore, allow an individualized approach accordingly to each subtype, with the subsequent improvement on patient outcomes.4,7
DiagnosisFirst and foremost, the diagnosis of MODY requires a high index of suspicion.59,60 In fact, MODY's diagnosis still remains a challenge, since this heterogeneous group of monogenic disorders comprises a large clinical spectrum and it usually overlaps with other types of diabetes. Consequently, many patients remain undiagnosed. Although MODY affects only a small part of diabetic subjects and its diagnosis is rare, it has important implications for the prognosis and treatment of patients, as well as for family members genetic counseling.24
The definitive diagnosis of MODY requires a molecular genetic study. These genetic tests are performed by direct sequencing of MODY genes (sensitivity >99%).24,60 The standard approach includes sequential screening of the three most common MODY genes: HNF-1α, GCK, and HNF-4α.61,62 However, with the advent of next-generation sequencing (NGS) technology, there has been a significant improvement in the speed and scalability of the genetic sequencing and an extended profile is also currently available.59,61
After a strong clinical suspicion, the genetic mutation diagnosis must be performed in order to accurately define MODY subtype and lead to the identification of other affected family members. Furthermore, the optimal treatment and risk for diabetes complications relies on the genetic defect.7,59
Although de novo mutations can arise, cascade screening of family members is essential to ensure those with diabetes get the correct diagnostic label and those at risk of inheriting the mutation are tested for diabetes and/or consider predictive genetic testing.16,59
Indications for genetic testing for MODY includes the following major diagnosis criteria widely accepted4,63:
- -
Hyperglycemia usually diagnosed before the age of 25 in at least 1 and ideally 2 family members;
- -
Autosomal dominant inheritance, with a vertical transmission of diabetes through at least 3 generations, and a similar phenotype shared by diabetic family members;
- -
Absence of insulin therapy at least 5 years after diagnosis or significant C-peptide levels even in a patient on insulin treatment;
- -
Insulin levels that are often in the normal range, although inappropriately low for the degree of hyperglycemia, suggesting a primary defect in β-cell function;
- -
Overweight or obesity is rarely associated (and is not required for the development of diabetes).
Notwithstanding, other remarkable MODY features encompasses: mild diabetes on presentation without significant ketosis; absence of pancreatic autoantibodies; glycosuria inappropriately observed along with euglycemia or mild hyperglycemia, in the absence of albuminuria and poorly controlled diabetes (low renal threshold that results in glycosuria is a typical MODY 3 feature); marked sensitivity to insulin secretagogues (sulfonylureas) in HNF-1α and HNF-4α subtypes; as well as no typical features of TIDM or T2DM.2,4,16
In order to achieve a more cost-effective approach, avoiding unnecessary genetic testing, a prediction model was developed to discriminate MODY patients from T1DM and T2DM, using logistic regression for that propose.64 The model, referred as The MODY Probability Calculator, offers a more standardized approach to select individuals for genetic testing. This online calculator (developed by Beverley Shields, University of Exeter; available at: https://www.diabetesgenes.org/mody-probability-calculator/), combines broad clinical information to predict the probability of testing positive for MODY and includes: age at diagnosis, body mass index, HbA1c level, therapy (insulin or OADs) and family history. When matched with traditional criteria, the prediction model improved the sensitivity and specificity for identifying MODY. However, this model was only validated in one European cohort of Caucasian individuals, lacking, therefore, a more generalized validation to other ethnicities.64
Differential diagnosisIn view of the high cost and limited availability of genetic testing, substantial efforts have recently been made to identify a specific, inexpensive and readily disposable non-genetic biomarker to discriminate between MODY and other types of diabetes. Along with the clinical features, it would aid in prioritizing patient for genetic testing (Table 2).7,59,65
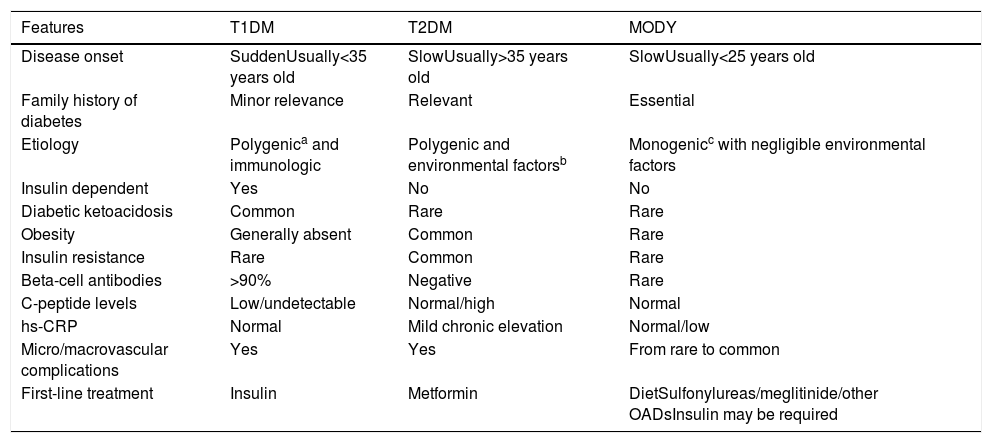
Differential diagnosis of distinctive forms of diabetes.
| Features | T1DM | T2DM | MODY |
|---|---|---|---|
| Disease onset | SuddenUsually<35 years old | SlowUsually>35 years old | SlowUsually<25 years old |
| Family history of diabetes | Minor relevance | Relevant | Essential |
| Etiology | Polygenica and immunologic | Polygenic and environmental factorsb | Monogenicc with negligible environmental factors |
| Insulin dependent | Yes | No | No |
| Diabetic ketoacidosis | Common | Rare | Rare |
| Obesity | Generally absent | Common | Rare |
| Insulin resistance | Rare | Common | Rare |
| Beta-cell antibodies | >90% | Negative | Rare |
| C-peptide levels | Low/undetectable | Normal/high | Normal |
| hs-CRP | Normal | Mild chronic elevation | Normal/low |
| Micro/macrovascular complications | Yes | Yes | From rare to common |
| First-line treatment | Insulin | Metformin | DietSulfonylureas/meglitinide/other OADsInsulin may be required |
T1DM, Type 1 Diabetes Mellitus; T2DM, Type 2 Diabetes Mellitus; hs-CRP, high sensitivity C-reactive protein.
It has been documented that in adults with diabetes for more than 5 years, the urinary C-peptide/creatinine ratio is higher in MODY 3 or MODY 1 when compared to T1DM (sensitivity 97% and specificity 96%).66 Furthermore, other biomarkers proposed for discrimination between MODY 3, MODY 1 and T2DM include 1,5-anhydroglucitol, the complement factors C5 and C8, apolipoprotein M and transthyretin, although none of them have enough specificity or sensitivity to be clinically.66 So far, one of the most promising biomarkers is high-sensitivity C-reactive protein (hs-CRP), which is significantly lower in MODY 3 than in TIDM, T2DM, MODY 2 and even in non-diabetic individuals.67,68
In fact, for most cases, the onset of MODY arises in childhood or adolescence, resembling T1DM. However, in these patients there is no complete lack of insulin and, therefore, insulin therapy will not be required, at least at an early stage of the disease. In some patients OADs or insulin therapy may be necessary, as a rapid progression of hyperglycemia can occur.12,16 The presence of autoantibodies for T1DM often precludes further testing for monogenic diabetes, since it makes MODY very unlikely, albeit positive autoantibodies in patients with monogenic diabetes has been reported.69 Thereby, a biomarker screening such as the combination of C-peptide and antibody screening (autoantibodies to GAD, insulin, the tyrosine phosphatases IA-2 and IA-2b, and ZnT8) should be performed prior to consideration of genetic testing for MODY.12,59 Actually, recent population-based studies have pointed toward this biomarker screening as an easy and cost-effective approach capable to discriminate patients with MODY.65
On the other hand, it is more difficult to differentiate between MODY and T2DM. The absence of insulin resistance features, particularly in adolescents with presumed T2DM, is suspicious for MODY, though no biochemical tests that reliably differentiate between these two diseases are currently available.14,16 Usually, MODY behaves in the same fashion as an adult T2DM but has some specific features in the glycemic control MODY.
ConclusionMODY is a genetically and clinically heterogeneous group of disorders. Its identification still remains a challenge for physicians, being largely underdiagnosed, though it has important implications for the individual and their families.
Recent advances on the genetic determinants and pathophysiology of MODY allowed a better understanding of its underlying molecular mechanisms and, therefore, have been very promising and insightful, providing us several profitable approaches in this field. Further studies will make possible to set up new therapeutic strategies, with drugs able to prevent, correct or at least delay the decline of pancreatic β-cell function. Moreover, with the sustained increase of genomic and metabolomic development, rapid screening tools for MODY mutations will become readily available and a self-directed assessment will afford for a more personalized treatment and, ultimately, for a better patient care.
Authors’ contributionAll the authors contributed equally to this work.
Funding sourcesThe authors have no funding to disclose related to this review.
Conflict of interestsThe authors declare no conflict of interests.
