




El síndrome de Cushing ectópico (SCE) es una entidad rara debida a la secreción de ACTH por tumores extrahipofisarios. Su baja frecuencia dificulta la adquisición de experiencia en su manejo. El objetivo de este trabajo es describir a los pacientes con SCE atendidos en el servicio de Endocrinología en un hospital de tercer nivel en un periodo de 15 años.
MétodosSe trata de un estudio retrospectivo de los datos clínicos, bioquímicos y radiológicos, tratamiento recibido, y evolución de los pacientes con SCE atendidos entre los años 2000 y 2015.
ResultadosSe incluyeron 9 pacientes (6 mujeres) con una edad media de 47 años. El síndrome clínico se desarrolló en un tiempo inferior a 3 meses en todos los casos excepto en uno, y la mayoría presentaba edemas, hiperpigmentación y/o hipopotasemia. La media del cortisol libre urinario y de la ACTH fue de 2.840μg/24h y 204pg/ml, respectivamente. El origen ectópico se confirmó por la combinación de pruebas dinámicas no invasivas y estudios radiológicos en la mayoría de los casos. El tumor responsable pudo identificarse en 8 casos y 7 presentaban diseminación metastásica. El tratamiento primario consistió en cirugía en un caso, cirugía más terapia sistémica en 3 y quimioterapia en otros 3. En 4 pacientes fue necesaria la suprarrenalectomía bilateral para controlar el hipercortisolismo. Tras un seguimiento medio de 40 meses, 3 habían fallecido, 5 permanecían vivos y en uno se había perdido el seguimiento.
ConclusionesSe confirma que el SCE abarca un amplio espectro de tumores de diferente agresividad y naturaleza. Habitualmente el origen ectópico del síndrome de Cushing puede sospecharse y confirmarse en la mayoría de los casos sin necesidad de pruebas invasivas. Tanto el control del hipercortisolismo como del tumor requieren múltiples modalidades terapéuticas, siendo recomendable el manejo multidisciplinar.
Ectopic Cushing's syndrome (ECS) is a rare condition caused by ACTH secretion by extrapituitary tumors. Its low frequency makes it difficult to acquire experience in its management. The aim of this study was to describe patients with ECS seen at the endocrinology department of a tertiary hospital over 15 years.
MethodsThis was a retrospective study of the clinical, biochemical and radiographic data, treatment, and course of patients with ECS seen from 2000 to 2015.
ResultsNine patients (6 of them female) with a mean age of 47 years were included in the study. The clinical syndrome developed in less than 3 months in all cases but one, and most patients also had edema, hyperpigmentation and/or hypokalemia. Mean urinary free cortisol and ACTH levels were 2,840μg/24h and 204pg/mL respectively. The ectopic origin was confirmed by a combination of dynamic non-invasive tests and radiographic studies in most cases. The tumor responsible could be identified in 8 cases, and 7 patients had metastatic dissemination. Primary treatment was surgery in one patient, surgery combined with systemic therapy in 3, and chemotherapy in the other 3 patients. Bilateral adrenalectomy was required in 4 patients to control hypercortisolism. After a mean follow-up of 40 months, 3 patients died, 5 were still alive, and one had been lost to follow-up.
ConclusionsOur study confirms that ECS covers a wide spectrum of tumors of different aggressiveness and nature. The ectopic origin of Cushing's syndrome can usually, be suspected and confirmed in most cases without the need for invasive tests. Control of both hypercortisolism and the tumor requires multiple treatment modalities, and multidisciplinary management is recommended.
La secreción de ACTH por tumores extrahipofisarios como causa de síndrome de Cushing (SC) fue descrita por Liddle en 1962, y es responsable de aproximadamente el 15% de los casos de SC endógeno1,2. La fuente de ACTH suele corresponder a neoplasias con diferenciación neuroendocrina de localización y agresividad muy variables. En el 60% de los casos el tumor es intratorácico (carcinoma microcítico de pulmón, carcinoide bronquial y tímico). El resto está constituido fundamentalmente por tumores neuroendocrinos (TNE) pancreáticos, feocromocitomas y carcinomas medulares de tiroides3–8.
La incidencia es similar en ambos sexos y la edad media de presentación es de 40-50 años, aunque también se han documentado casos en niños y adolescentes9. El espectro de presentación clínica es amplio y abarca desde formas fulminantes de hipercortisolismo, en las que predominan la miopatía, los edemas, la hiperpigmentación y las alteraciones psiquiátricas, a otras formas de curso más indolente con síntomas similares a los típicos de la enfermedad de Cushing (EC). En este último caso establecer el diagnóstico diferencial entre SC ectópico (SCE) y EC constituye un verdadero reto3. Asimismo, la identificación de la fuente de ACTH y el tratamiento del hipercortisolismo ofrecen a menudo considerables dificultades.
Describimos nuestra experiencia en el diagnóstico, tratamiento y pronóstico a largo plazo de los pacientes con SCE atendidos en nuestro centro a lo largo de un periodo de 15 años.
Material y métodosPacientesSe incluyeron en el estudio los pacientes con SCE atendidos en el Servicio de Endocrinología del Hospital Puerta de Hierro de Majadahonda entre los años 2000 y 2015. Se consideraron portadores de un SCE los pacientes con SC ACTH dependiente que además reunieran alguno de los siguientes criterios indicativos de secreción extrahipofisaria de ACTH: a) cuadro clínico y bioquímico sugestivo y evidencia de tumor con alta sospecha de TNE; b) 2 test dinámicos compatibles y evidencia de TNE; c) cateterismo de senos petrosos inferiores (CSPI) compatible.
Estudio bioquímicoIncluyó en todos los casos la determinación, en al menos 2 ocasiones, de cortisol sérico (CS) a las 8:00h (valor normal [VN]: 4,3-22,4μg/dl), ACTH basal (8:00h) (VN: 9-55 pg/ml) y cortisol libre en orina de 24h (CLU) (VN: 11-71μg/24h). Dependiendo de las dificultades diagnósticas, en algunos casos se recurrió a uno o más de los siguientes test: CS nocturno (23:00h) (VN<7,5μg/dl), test de supresión fuerte con dexametasona (TSFD) (2mg de dexametasona/6h durante 2 días), test de CRH, y CSPI. Se definió supresión en el TSFD como el descenso del CLU>90% y/o del CS>50% respecto al valor basal. El test de CRH se consideró positivo para EC en caso de incremento de ACTH>50% y/o de CS>20% respecto al valor basal. En el CSPI un gradiente central/periférico de ACTH<2 en situación basal y <3 tras CRH se consideró indicativo de secreción ectópica de ACTH.
El CS y la ACTH se determinaron mediante quimioluminiscencia (Advia Centaur XP, Siemens Healthineers, Erlangen, Alemania). El CLU se determinó mediante radioinmunoanálisis (Stratec, Diasource, Louvain-la-Neuve, Bélgica).
Estudios de imagenSe realizó tomografía computarizada (TC) o resonancia magnética (RM) en todos los pacientes. Adicionalmente, en algunos casos se emplearon uno o más de los siguientes procedimientos: gammagrafía con 111In DTPA-octreótido (octreoscan), gammagrafía con 123I-metayodobencilguanidina (MIBG), PET con fluorodesoxiglucosa (18FDG) y PET con fluorodopa (18F-DOPA).
Otras exploraciones efectuadas con el objetivo de identificar la fuente de ACTH o confirmar la sospecha de TNE fueron: a) Cuantificación de marcadores tumorales: cromogranina A, gastrina, insulina, glucagón, péptido intestinal vasoactivo, somatostatina, ácido 5-hidroxiindolacético, antígeno carcinoembrionario, calcitonina y metanefrinas fraccionadas en orina de 24h, b) exploraciones endoscópicas digestivas y/o bronquiales y c) toma de muestras histológicas.
ResultadosEntre los años 2000 y 2015 se identificaron 9 casos de SCE. De ellos 6 fueron mujeres, y la edad media al diagnóstico fue de 47 años (tabla 1).
Presentación clínica
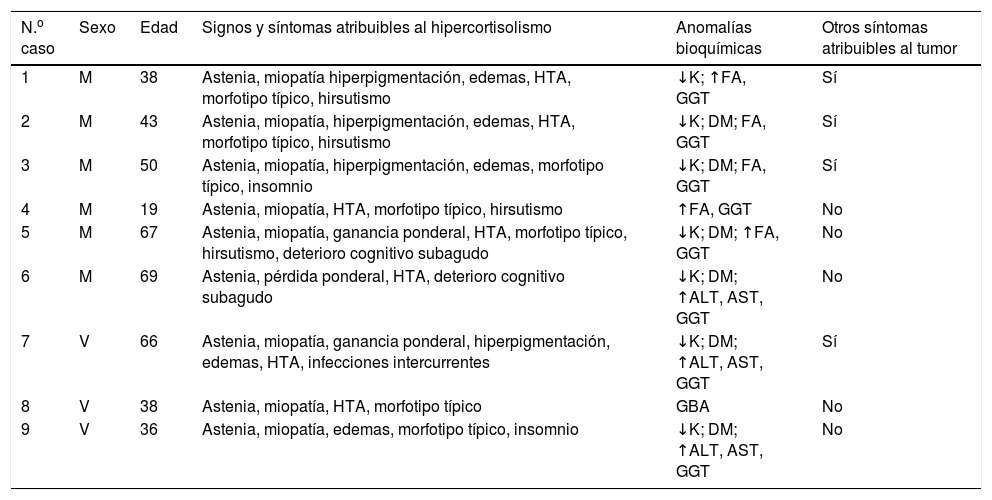
| N.o caso | Sexo | Edad | Signos y síntomas atribuibles al hipercortisolismo | Anomalías bioquímicas | Otros síntomas atribuibles al tumor |
|---|---|---|---|---|---|
| 1 | M | 38 | Astenia, miopatía hiperpigmentación, edemas, HTA, morfotipo típico, hirsutismo | ↓K; ↑FA, GGT | Sí |
| 2 | M | 43 | Astenia, miopatía, hiperpigmentación, edemas, HTA, morfotipo típico, hirsutismo | ↓K; DM; FA, GGT | Sí |
| 3 | M | 50 | Astenia, miopatía, hiperpigmentación, edemas, morfotipo típico, insomnio | ↓K; DM; FA, GGT | Sí |
| 4 | M | 19 | Astenia, miopatía, HTA, morfotipo típico, hirsutismo | ↑FA, GGT | No |
| 5 | M | 67 | Astenia, miopatía, ganancia ponderal, HTA, morfotipo típico, hirsutismo, deterioro cognitivo subagudo | ↓K; DM; ↑FA, GGT | No |
| 6 | M | 69 | Astenia, pérdida ponderal, HTA, deterioro cognitivo subagudo | ↓K; DM; ↑ALT, AST, GGT | No |
| 7 | V | 66 | Astenia, miopatía, ganancia ponderal, hiperpigmentación, edemas, HTA, infecciones intercurrentes | ↓K; DM; ↑ALT, AST, GGT | Sí |
| 8 | V | 38 | Astenia, miopatía, HTA, morfotipo típico | GBA | No |
| 9 | V | 36 | Astenia, miopatía, edemas, morfotipo típico, insomnio | ↓K; DM; ↑ALT, AST, GGT | No |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; DM: diabetes; FA: fosfatasa alcalina; GBA: glucemia basal alterada; GGT: gamma glutamiltransferasa; HTA: hipertensión arterial; K: potasio sérico; M: mujer; V: varón.
Al diagnóstico, los síntomas más frecuentes fueron astenia y debilidad muscular como expresión de miopatía proximal (tabla 1). Siete pacientes referían además HTA y aumento de peso, 4 tenían hiperpigmentación, edemas, hirsutismo y alteraciones neurocognitivas o insomnio, y un caso presentó infecciones oportunistas resistentes al tratamiento. Todos los pacientes salvo 2 casos (6, 7) tenían un morfotipo característico con distribución centrípeta de la grasa, además 2 de ellos presentaban estrías cutáneas y/o equimosis (casos 8, 9). Todos los casos desarrollaron un SC florido y todos excepto uno (caso 8) lo hicieron en un tiempo inferior a 3 meses.
La glucemia basal media fue de 185mg/dl (84-344) y se encontraba en rango de diabetes en 6 pacientes, 2 de los cuales (casos 5 y 7) tenían una diabetes conocida previamente. Siete tenían hipopotasemia (valor medio: 2,68mmol/l [rango 2-3,1]) y 8 presentaban hipertransaminasemia (patrón de colestasis disociada en 5 y mixto en 3).
En el momento de la presentación ningún caso había sido diagnosticado de un proceso neoplásico, si bien 4 de los 9 casos presentaban síntomas atribuibles al mismo, distintos de los propios del SC (síndrome de Zollinger-Ellison en 3 casos y disnea en otro).
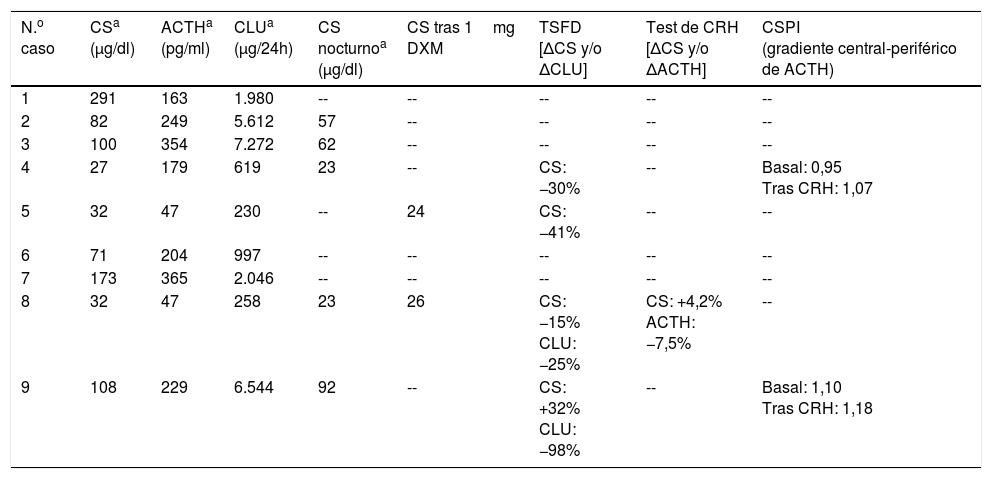
Diagnóstico bioquímicoEl valor medio de CS basal fue 102μg/dl y el de CLU 2.840μg/24h (tabla 2). En todos los casos el CLU superó en más de 3 veces el límite superior de la normalidad, con una media de 20 veces (rango 4,5-100 veces). El CS nocturno medio (n=5) fue 51μg/dl y el CS tras 1mg DXM (n=2) 25μg/dl. El nivel medio de ACTH fue de 204pg/ml, con valores dentro del rango normal en solo 2 de los 9 casos.
Parámetros hormonales
| N.o caso | CSa (μg/dl) | ACTHa (pg/ml) | CLUa (μg/24h) | CS nocturnoa (μg/dl) | CS tras 1mg DXM | TSFD [ΔCS y/o ΔCLU] | Test de CRH [ΔCS y/o ΔACTH] | CSPI (gradiente central-periférico de ACTH) |
|---|---|---|---|---|---|---|---|---|
| 1 | 291 | 163 | 1.980 | -- | -- | -- | -- | -- |
| 2 | 82 | 249 | 5.612 | 57 | -- | -- | -- | -- |
| 3 | 100 | 354 | 7.272 | 62 | -- | -- | -- | -- |
| 4 | 27 | 179 | 619 | 23 | -- | CS: −30% | -- | Basal: 0,95 Tras CRH: 1,07 |
| 5 | 32 | 47 | 230 | -- | 24 | CS: −41% | -- | -- |
| 6 | 71 | 204 | 997 | -- | -- | -- | -- | -- |
| 7 | 173 | 365 | 2.046 | -- | -- | -- | -- | -- |
| 8 | 32 | 47 | 258 | 23 | 26 | CS: −15% CLU: −25% | CS: +4,2% ACTH: −7,5% | -- |
| 9 | 108 | 229 | 6.544 | 92 | -- | CS: +32% CLU: −98% | -- | Basal: 1,10 Tras CRH: 1,18 |
CLU: cortisol libre urinario; CS: cortisol sérico; CSPI: cateterismo de senos petrosos inferiores; TSFD: test de supresión fuerte con dexametasona.
Valores normales: CS (4,3-22,4μg/dl), ACTH (9-55pg/ml), CLU (11-71μg/24h), CS nocturno (<7,5μg/dl), CS tras 1mg DXM (<1,8μg/dl).
En 5 casos (1, 2, 3, 6 y 7) el origen ectópico se estableció por los niveles de CLU y ACTH marcadamente elevados, junto a la evidencia radiológica de patología tumoral potencialmente responsable del cuadro. En los 4 casos restantes se practicó TSFD como primera aproximación al diagnóstico diferencial entre EC y SCE. En 3 de ellos (4, 5 y 8), todos con CLU moderadamente elevado, se constató ausencia de supresión del CS y/o CLU. El origen ectópico del SC se dio por demostrado por el hallazgo de metástasis de TNE en un caso (5) por test de CRH negativo y demostración de TNE en otro caso (8) y por CSPI con ausencia de gradiente en el tercero (4). El cuarto paciente sometido a TSFD (9), cuyos niveles de CLU eran marcadamente elevados, mostró supresión del CLU>90%. La RM hipofisaria evidenció una dudosa lesión pero el CSPI puso de manifiesto el origen extrahipofisario de la secreción de ACTH.
Localización de la fuente de ACTHEn todos los casos se realizaron pruebas de imagen (TC o RM). En 2 de ellos previo al diagnóstico de SCE (6 y 8) y en el resto para localizar la fuente de secreción ectópica de ACTH.
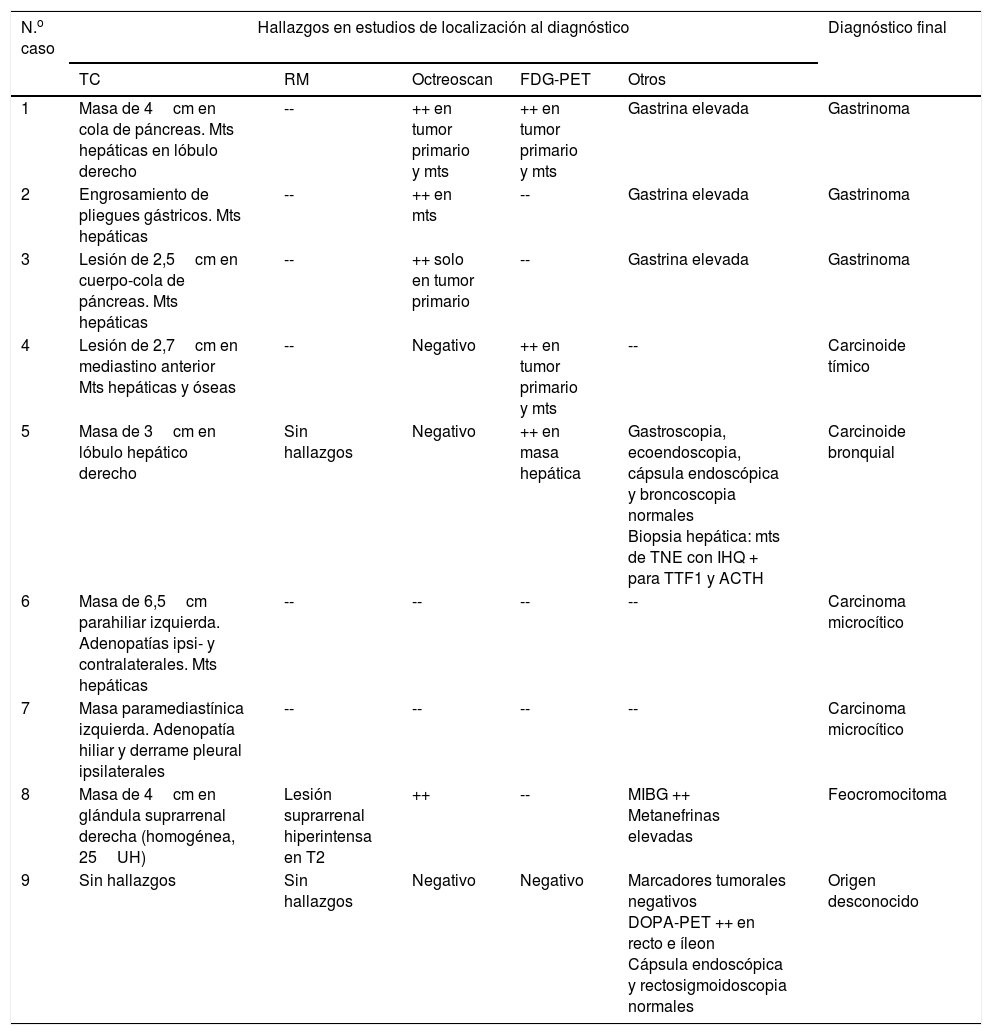
En los que se realizó el estudio tras confirmarse el diagnóstico de SCE, en 4 casos (1, 3, 4 y 7) se pudo identificar una lesión potencialmente causante del SCE, con diseminación metastásica en todos ellos (tabla 3). En un caso (2) se identificaron metástasis hepáticas múltiples y un engrosamiento de pliegues gástricos que junto a los datos clínicos y bioquímicos (diarrea y gastrina>22.000pg/ml [VN<100]) confirmaban el diagnóstico de gastrinoma. En otro caso (5) se evidenció una lesión hepática única cuya biopsia demostró que se trataba de una metástasis de TNE con inmunohistoquímica positiva para ACTH. En un último caso (9) las pruebas de imagen no lograron identificar ninguna lesión
Resultados de los estudios de localización iniciales
| N.o caso | Hallazgos en estudios de localización al diagnóstico | Diagnóstico final | ||||
|---|---|---|---|---|---|---|
| TC | RM | Octreoscan | FDG-PET | Otros | ||
| 1 | Masa de 4cm en cola de páncreas. Mts hepáticas en lóbulo derecho | -- | ++ en tumor primario y mts | ++ en tumor primario y mts | Gastrina elevada | Gastrinoma |
| 2 | Engrosamiento de pliegues gástricos. Mts hepáticas | -- | ++ en mts | -- | Gastrina elevada | Gastrinoma |
| 3 | Lesión de 2,5cm en cuerpo-cola de páncreas. Mts hepáticas | -- | ++ solo en tumor primario | -- | Gastrina elevada | Gastrinoma |
| 4 | Lesión de 2,7cm en mediastino anterior Mts hepáticas y óseas | -- | Negativo | ++ en tumor primario y mts | -- | Carcinoide tímico |
| 5 | Masa de 3cm en lóbulo hepático derecho | Sin hallazgos | Negativo | ++ en masa hepática | Gastroscopia, ecoendoscopia, cápsula endoscópica y broncoscopia normales Biopsia hepática: mts de TNE con IHQ + para TTF1 y ACTH | Carcinoide bronquial |
| 6 | Masa de 6,5cm parahiliar izquierda. Adenopatías ipsi- y contralaterales. Mts hepáticas | -- | -- | -- | -- | Carcinoma microcítico |
| 7 | Masa paramediastínica izquierda. Adenopatía hiliar y derrame pleural ipsilaterales | -- | -- | -- | -- | Carcinoma microcítico |
| 8 | Masa de 4cm en glándula suprarrenal derecha (homogénea, 25UH) | Lesión suprarrenal hiperintensa en T2 | ++ | -- | MIBG ++ Metanefrinas elevadas | Feocromocitoma |
| 9 | Sin hallazgos | Sin hallazgos | Negativo | Negativo | Marcadores tumorales negativos DOPA-PET ++ en recto e íleon Cápsula endoscópica y rectosigmoidoscopia normales | Origen desconocido |
IHQ: inmunohistoquímica; MIBG: metayodobencilguanidina; Mts: metástasis; RM: resonancia magnética; TC: tomografía computarizada; TNE: tumor neuroendocrino; TTF1: thyroid transcription factor 1; UH: unidades hounsfield.
En 2 pacientes se habían realizado las pruebas de imagen antes de confirmarse el diagnóstico de SCE (6, 8). En el caso 6, el estudio (motivado por una hipertransaminasemia) evidenció una masa pulmonar con metástasis hepáticas. En el caso 8, una TC abdominal realizada durante la investigación del hipercortisolismo en otro centro identificó una masa adrenal indeterminada de la que no se había realizado más estudio. Tras el diagnóstico de SCE en nuestro centro, el estudio de la masa permitió catalogarla como un feocromocitoma.
Se practicó octreoscan en los 3 casos sin evidencia de tumor primario en los estudios de imagen anatómicos (2, 5 y 9), pero no aportó información adicional en ningún caso. Se recurrió a FDG-PET en 2 casos: en uno (5) se identificó la metástasis hepática ya conocida pero no el tumor primario, y en el otro (9) no se evidenció ninguna lesión. En este último caso se practicó PET-18F-DOPA, que mostró captación patológica en recto e íleon. Sin embargo, la rectosigmoidoscopia y la cápsula endoscópica no detectaron ninguna lesión.
Asimismo, se practicó octreoscan en 4 de los 6 casos localizados previamente mediante TC/RM. El resultado fue positivo en 3 (1, 3 y 8) y negativo en uno (4).
En conjunto, las pruebas de localización iniciales condujeron a la identificación del tumor primario en 6 casos, en 2 casos permitieron identificar metástasis pero no el tumor primario (2 y 5) y en un caso no consiguieron identificar ninguna lesión (9). La repetición de estudios de imagen a lo largo del seguimiento permitió identificar el tumor primario (nódulo pulmonar de 0,9cm) 18 meses después del diagnóstico inicial en el caso 5. Por el contrario, en el caso 9 la repetición de estudios de imagen y marcadores tumorales no lograron identificar el tumor primario, que actualmente permanece oculto (tabla 3).
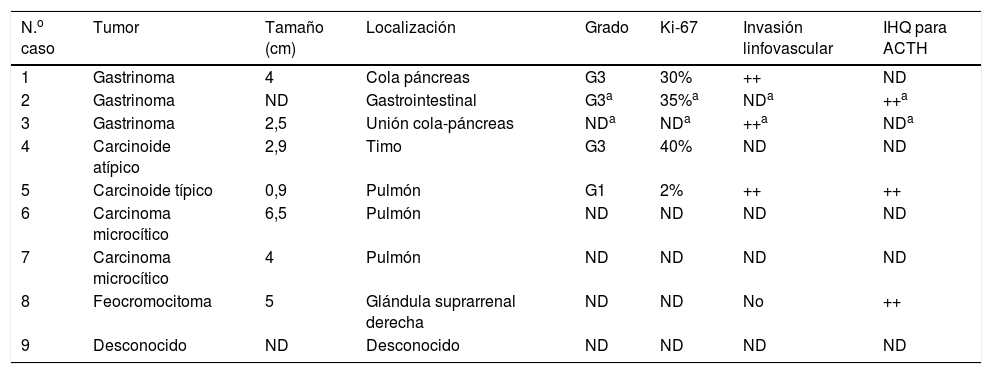
Características del tumor primarioSe identificó el tumor primario en 7 casos: 2 gastrinomas pancreáticos, 2 carcinomas microcíticos de pulmón, un carcinoide tímico, un carcinoide bronquial y un feocromocitoma. En el caso 2 se estableció el diagnóstico de gastrinoma, sin identificarse la localización exacta. Las localizaciones más frecuentes fueron gastrointestinal (3 casos) y pulmonar (3 casos). El tamaño del tumor primario osciló entre 0,9-6,5cm, y en 7 casos se encontraban diseminados al diagnóstico. Se efectuó inmunohistoquímica para ACTH en 3 casos y resultó positiva en todos ellos (tabla 4).
Características del tumor primario
| N.o caso | Tumor | Tamaño (cm) | Localización | Grado | Ki-67 | Invasión linfovascular | IHQ para ACTH |
|---|---|---|---|---|---|---|---|
| 1 | Gastrinoma | 4 | Cola páncreas | G3 | 30% | ++ | ND |
| 2 | Gastrinoma | ND | Gastrointestinal | G3a | 35%a | NDa | ++a |
| 3 | Gastrinoma | 2,5 | Unión cola-páncreas | NDa | NDa | ++a | NDa |
| 4 | Carcinoide atípico | 2,9 | Timo | G3 | 40% | ND | ND |
| 5 | Carcinoide típico | 0,9 | Pulmón | G1 | 2% | ++ | ++ |
| 6 | Carcinoma microcítico | 6,5 | Pulmón | ND | ND | ND | ND |
| 7 | Carcinoma microcítico | 4 | Pulmón | ND | ND | ND | ND |
| 8 | Feocromocitoma | 5 | Glándula suprarrenal derecha | ND | ND | No | ++ |
| 9 | Desconocido | ND | Desconocido | ND | ND | ND | ND |
IHQ: inmunohistoquímica; ND: no disponible.
La identificación definitiva de la fuente de ACTH se estableció en 4 casos, bien por la demostración de tinción positiva para ACTH en la inmunohistoquímica (2 y 5), bien por la resolución del hipercortisolismo tras el tratamiento exitoso del tumor (1), o por ambos (8).
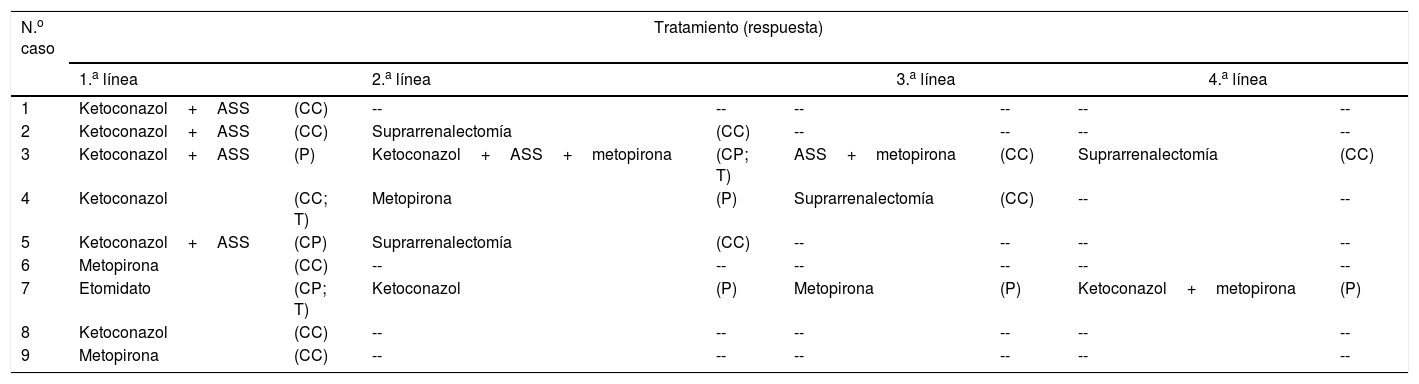
TratamientoTratamiento del hipercortisolismoTodos los pacientes recibieron tratamiento médico para el hipercortisolismo antes de someterse a tratamiento antitumoral (tabla 5).
- a)
En 6 casos se empleó ketoconazol como primera medida para el control del hipercortisolismo, en 2 de ellos en monoterapia y en los 4 restantes combinado con análogos de somatostatina (ASS) (lanreotide u octreótido). Los 2 casos tratados con monoterapia (4 y 8) presentaban una elevación moderada del CLU y ambos lograron normalizarlo con el tratamiento. De los 4 casos tratados con doble terapia (ketoconazol+ASS) el CLU se normalizó en 2 casos (1 y 2) y persistió elevado en otros 2 (3 y 5), uno de los cuales presentaba niveles de CLU solo ligeramente elevados (5).
- b)
En 2 casos (1 y 9) se empleó metopirona como tratamiento inicial del hipercortisolismo debido a una elevación preexistente de las transaminasas que impidió el uso de ketoconazol. En ambos pacientes se logró control del hipercortisolismo de forma mantenida pese a persistencia (9) o recidiva (1) tumoral.
- c)
Por último, en un caso (7) el tratamiento inicial del hipercortisolismo se llevó a cabo con etomidato. En este caso, el paciente se hallaba ingresado en la Unidad de Cuidados Intensivos y con intubación orotraqueal debido a las complicaciones derivadas del hipercortisolismo, por lo que se precisaba un tratamiento de administración intravenosa y capaz de lograr un rápido control del hipercortisolismo que amenazaba la vida del paciente. Se empleó etomidato en pauta bloqueo-sustitución, lográndose un descenso significativo de los niveles de CS (de 471 a 149μg/dl), pero el tratamiento se suspendió al sexto día debido a una marcada elevación de la osmolaridad plasmática atribuible al propilenglicol empleado como excipiente.
Tratamiento del hipercortisolismo
| N.o caso | Tratamiento (respuesta) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1.a línea | 2.a línea | 3.a línea | 4.a línea | |||||
| 1 | Ketoconazol+ASS | (CC) | -- | -- | -- | -- | -- | -- |
| 2 | Ketoconazol+ASS | (CC) | Suprarrenalectomía | (CC) | -- | -- | -- | -- |
| 3 | Ketoconazol+ASS | (P) | Ketoconazol+ASS+metopirona | (CP; T) | ASS+metopirona | (CC) | Suprarrenalectomía | (CC) |
| 4 | Ketoconazol | (CC; T) | Metopirona | (P) | Suprarrenalectomía | (CC) | -- | -- |
| 5 | Ketoconazol+ASS | (CP) | Suprarrenalectomía | (CC) | -- | -- | -- | -- |
| 6 | Metopirona | (CC) | -- | -- | -- | -- | -- | -- |
| 7 | Etomidato | (CP; T) | Ketoconazol | (P) | Metopirona | (P) | Ketoconazol+metopirona | (P) |
| 8 | Ketoconazol | (CC) | -- | -- | -- | -- | -- | -- |
| 9 | Metopirona | (CC) | -- | -- | -- | -- | -- | -- |
ASS: análogos de somatostatina; CC: control completo; CP: control parcial; P: persistencia; T: toxicidad.
En 5 casos fue necesario recurrir a una segunda línea de tratamiento, bien por persistencia o recidiva del hipercortisolismo, bien por desarrollo de toxicidad. En 2 de estos casos (2 y 5) la terapia elegida fue la suprarrenalectomía bilateral (SR) y en los otros 3 se utilizó un fármaco alternativo o bien la asociación de varios. Finalmente, 2 de estos 3 casos (3 y 4) fueron sometidos a SR, por dificultades en el control del hipercortisolismo y para evitar la exposición prolongada a inhibidores de esteroidogénesis. El tercer caso, tratado con etomidato en primera línea, falleció en pocos días sin haber alcanzado en ningún momento el control del hipercortisolismo
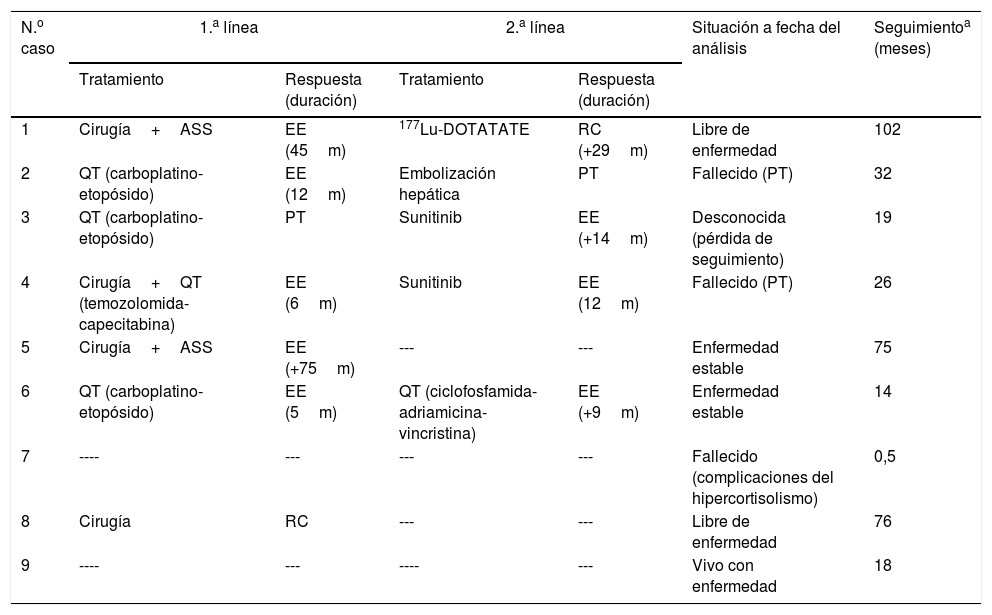
Tratamiento antitumoralPuesto que la mayoría de los casos se hallaban diseminados en el momento del diagnóstico, solo se pudo efectuar cirugía con intención curativa en uno (8), que alcanzó de este modo la remisión del SC. De los 7 casos con diseminación metastásica (tabla 6):
- a)
Tres se sometieron a cirugía: en un caso (4) solo se efectuó resección del tumor primario con intención de facilitar el control hormonal y disponer de confirmación histológica; en otro caso (5) se efectuó resección completa de la metástasis hepática (única) en un primer tiempo y del tumor primario en un segundo tiempo dado que trascurrieron 18 meses entre la identificación de una y otro; y en el tercer caso (1) se resecaron simultáneamente el tumor primario y las metástasis (hepatectomía derecha). Tras la cirugía, en un caso (4) se empleó quimioterapia (QT) (temozolomida+capecitabina) para el control tumoral lográndose enfermedad estable (EE) durante 6 meses; en los otros 2 casos (1 y5) se emplearon ASS lográndose EE durante 45 y 75 meses, respectivamente.
- b)
En 3 casos se empleó QT mediante esquema carboplatino-etopósido como tratamiento primario de la enfermedad. En un caso (3) el tratamiento se suspendió tras 3 ciclos al evidenciarse progresión tumoral (PT), y en 2 casos (2 y 6) se logró EE durante 12 y 5 meses tras haber recibido un total de 6 y 4 ciclos respectivamente.
- c)
En un caso (7) el curso de la enfermedad fue altamente agresivo y condicionó un rápido y profundo deterioro clínico que hizo inviable el tratamiento antitumoral y el paciente falleció 13 días después del diagnóstico.
Tratamiento antitumoral y situación a fecha del análisis
| N.o caso | 1.a línea | 2.a línea | Situación a fecha del análisis | Seguimientoa (meses) | ||
|---|---|---|---|---|---|---|
| Tratamiento | Respuesta (duración) | Tratamiento | Respuesta (duración) | |||
| 1 | Cirugía+ASS | EE (45m) | 177Lu-DOTATATE | RC (+29m) | Libre de enfermedad | 102 |
| 2 | QT (carboplatino-etopósido) | EE (12m) | Embolización hepática | PT | Fallecido (PT) | 32 |
| 3 | QT (carboplatino-etopósido) | PT | Sunitinib | EE (+14m) | Desconocida (pérdida de seguimiento) | 19 |
| 4 | Cirugía+QT (temozolomida-capecitabina) | EE (6m) | Sunitinib | EE (12m) | Fallecido (PT) | 26 |
| 5 | Cirugía+ASS | EE (+75m) | --- | --- | Enfermedad estable | 75 |
| 6 | QT (carboplatino-etopósido) | EE (5m) | QT (ciclofosfamida-adriamicina-vincristina) | EE (+9m) | Enfermedad estable | 14 |
| 7 | ---- | --- | --- | --- | Fallecido (complicaciones del hipercortisolismo) | 0,5 |
| 8 | Cirugía | RC | --- | --- | Libre de enfermedad | 76 |
| 9 | ---- | --- | ---- | --- | Vivo con enfermedad | 18 |
ASS: análogos de somatostatina; EE: enfermedad estable; PT: progresión tumoral; QT: quimioterapia; RC: remisión completa; SCE: síndrome de Cushing ectópico.
Se evidenció PT en 4 de los 5 pacientes que habían obtenido EE con el tratamiento primario, con un tiempo medio hasta la progresión de 17 meses. Tras la progresión, uno de ellos (1) recibió tratamiento con 177Lu-DOTATATE, alcanzando remisión completa de la enfermedad y permanecía en dicha situación a fecha de la última evaluación (29 meses tras la finalización del tratamiento). En otro caso (2) se practicó embolización de las lesiones metastásicas en ambos lóbulos hepáticos, a pesar de lo cual presentó progresión de la enfermedad y falleció a los 32 meses del diagnóstico por insuficiencia hepática. El tercero (4) fue tratado con sunitinib consiguiendo EE durante 12 meses. Por último, el cuarto caso (6) recibió una segunda línea de QT con ciclofosfamida+adriamicina+vincristina, manteniendo EE a fecha de la última evaluación (9 meses tras la finalización del tratamiento).
El paciente que presentó PT con el tratamiento primario (3) recibió sunitinib como segunda línea de tratamiento logrando EE durante 14 meses, transcurridos los cuales se perdió el seguimiento.
Situación al final del seguimientoTras un seguimiento medio de 40 meses, 2 pacientes habían fallecido por PT a los 32 y 26 meses del diagnóstico respectivamente y uno por complicaciones del hipercortisolismo a los 13 días del diagnóstico, 2 se encontraban en RC, 3 permanecían estables, y en uno se había perdido el seguimiento (tabla 6).
DiscusiónEl SCE es una entidad infrecuente, tal como lo atestigua la presente serie, que ha logrado reunir 9 casos a lo largo de 15 años en un centro con un área poblacional de referencia de más de 450.000 habitantes y con potencial atracción de casos de otras áreas por su reconocida experiencia en el diagnóstico y tratamiento del SC. No obstante, esta es la serie más numerosa publicada en nuestro país hasta la fecha.
La edad y distribución por sexos fueron similares a las descritas previamente. Clínicamente se pudieron identificar los 2 extremos del espectro clínico del SCE: enfermedad lentamente progresiva, indistinguibles del SC de origen hipofisario (caso 8) y enfermedad de curso explosivo manifestada con síntomas constitucionales y ausencia del morfotipo clásico (caso 7). Sin embargo, la mayoría de los pacientes presentaron datos que permitían sospechar una secreción ectópica de ACTH tales como edemas o hipopotasemia. Estos signos reflejan la existencia de un cortisol marcadamente elevado, capaz de acceder al receptor de mineralcorticoides del túbulo renal al sobrepasar la capacidad inactivadora de la enzima β-hidroxiesteroide-deshidrogenasa tipo 2 y, aunque no son exclusivos del SCE, son muy infrecuentes en el SC de otro origen5,10–14.
Seis de los 9 casos tenían niveles de CS, CLU y ACTH marcadamente superiores a los habituales en la EC, y un caso más presentaba una ACTH claramente elevada. Solo 2 casos tenían niveles de CS, CLU y ACTH en rango similar a los de la EC (no obstante, se debe tener en cuenta el considerable solapamiento que existe en los valores de estas determinaciones en los pacientes con SCE y EC) y ambos carecían de datos clínicos sugestivos de SCE. De hecho uno de estos 2 pacientes (descrito en detalle en otra publicación)15 había sido diagnosticado erróneamente de EC y sometido a cirugía hipofisaria antes de acudir a nuestro centro. Los niveles de ACTH en el paciente con tumor oculto superaron a los observados en 4 casos con tumor evidente, lo que confirma la ausencia de correlación entre los valores de ACTH y el tamaño tumoral demostrada en otros estudios14.
El solapamiento de datos clínicos y hormonales entre la EC y el SCE, descrito hasta en un 30% de los casos, obliga a recurrir a test adicionales para diferenciar ambas entidades1,10,12,16–18. El TSFD es altamente específico para identificar a los sujetos con EC cuando la supresión de CLU es superior al 90% pero la ausencia de supresión es poco específica de SCE19. El CSPI tiene una sensibilidad y especificidad muy altas y es la prueba de elección empleada en muchos centros pero se trata de un procedimiento invasivo no exento de riesgos, por lo que debería reservarse para aquellos casos con datos clínicos y/o bioquímicos sospechosos o con test no invasivos compatibles con origen ectópico en los que no exista evidencia de lesión tumoral en las pruebas radiológicas20–22. Sin embargo, con frecuencia, en el SCE, dada la gravedad de la situación clínica que obliga a una actitud terapéutica temprana y los riesgos asociados a algunas de estas pruebas, estos estudios no se llegan a realizar, tal y como se describe en nuestra serie.
A excepción del carcinoma medular de tiroides, todas las estirpes tumorales asociadas con secreción ectópica de ACTH se encuentran representadas en nuestra serie a pesar de su limitado tamaño. Aunque el carcinoma microcítico de pulmón y el carcinoide bronquial suelen ser los más frecuentes5,6,10–12, el gastrinoma fue el más prevalente en nuestro estudio, afectando a 3 de los 9 pacientes, frente a aproximadamente un 15% observado en la mayor parte de las series. Esta diferencia probablemente sea resultado del azar pues no hemos encontrado otra razón que la justifique.
En 8 casos (89%) pudo identificarse el tumor o sus metástasis mediante pruebas de imagen convencionales efectuadas durante o poco después del diagnóstico del SCE. Esta tasa de localización es superior a la reportada en otras series, que oscila en torno al 50%23, pero hay que tener en cuenta que en nuestra serie casi la mitad de los pacientes presentaban síntomas distintos a los derivados del SC (diarrea y disnea) y la mayoría de los casos mostraban un estadio tumoral avanzado. La gammagrafía con ASS ocasionalmente puede ser de ayuda para la identificación de tumores no localizados con TC/RM5,23–26, pero no sucedió así en el único caso de nuestra serie sin evidencia de enfermedad tumoral mediante pruebas de imagen anatómicas. Como suele ser habitual, tampoco el PET-FDG aportó información a este respecto dado que los tumores ocultos suelen presentar una tasa metabólica baja26. El PET con 18F-DOPA, un radiotrazador más específico de TNE, mostró captación en íleon y recto pero los estudios endoscópicos no evidenciaron anomalías en estas localizaciones. Es probable, por tanto, que se tratase de un falso positivo del 18F-DOPA PET, si bien la especificidad documentada hasta ahora para este procedimiento ha sido del 100%23. Los marcadores tumorales (gastrina, calcitonina, metanefrinas), que pueden ayudan a localizar la fuente de ACTH1–3,5, tampoco fueron de utilidad en esta ocasión. La fuente de ACTH permanece, pues, desconocida en este caso, hecho que sucede aproximadamente en el 15% de los SCE5,11,24,27. A pesar de que un intento repetidamente fallido para encontrar el tumor indica un pronóstico favorable, no garantiza la ausencia de un proceso maligno4,5,20,21 por lo que el seguimiento a largo plazo es obligado en estos casos.
La relación temporal entre el diagnóstico del SCE y el del tumor es variable, pero es habitual que el SCE se manifieste en el contexto de progresión o recaída tumoral1–5,12,16. En consonancia con este hecho, 7 de los 9 casos recogidos en la presente serie presentaban diseminación metastásica al diagnóstico. Sin embargo, ningún paciente había sido diagnosticado de un proceso neoplásico cuando se estableció la sospecha de SC. Este hecho probablemente se deba a la baja especificidad de los síntomas que impide que se reconozca su origen tumoral tal como sucedió en los 3 gastrinomas. En ellos el inicio del síndrome de Zollinger-Ellison tuvo lugar al menos un año antes del inicio del SC.
El único tratamiento curativo del SCE es la resección o eliminación completa de la neoplasia, pero dada la frecuente diseminación metastásica en nuestra serie solo se logró en 2 de los 9 casos, una tasa similar al 10-15% recogido en la literatura28.
En tumores pobremente diferenciados, la alternativa de elección es la QT basada en platino, mientras que en tumores bien diferenciados han demostrado utilidad los ASS, la QT, los inhibidores de mTOR (everolimus) o de tirosincinasas (sunitinib), y la terapia con radionúclidos, con tasas de respuesta variables29. La evidencia disponible actualmente no ha permitido establecer cuál es el tratamiento inicial de elección ni la secuencia de tratamientos más apropiada tras la PT. Recientemente el tratamiento con radionúclidos (177Lu-DOTATATE) en sujetos con TNE de intestino medio irresecable y en progresión a ASS ha demostrado una tasa de respuesta radiológica y una duración de la misma superior a cualquier otra modalidad terapéutica30. De acuerdo con esta evidencia, el único caso de nuestra serie tratado con 177Lu-DOTATATE logró una respuesta clínica y radiológica completa y mantenida en el tiempo.
El control del hipercortisolismo mediante tratamiento médico es obligado antes de someter al paciente a cirugía o QT pues disminuye de forma significativa la morbimortalidad y la toxicidad relacionada con dichas terapias, e incrementa la tasa de respuesta31. Por otra parte, constituye un elemento clave en el control del SCE cuando no es posible la remisión tumoral. El ketoconazol y la metopirona (asociados en ocasiones a ASS) fueron eficaces en la mayoría de nuestros casos.
Por su efecto casi inmediato y la posibilidad de administración intravenosa, el etomidato debe reservarse para situaciones críticas derivadas de un hipercortisolismo amenazante para la vida32. La hiperosmolaridad asociada al tratamiento con etomidato es un marcador subrogado de las concentraciones de propilenglicol33, usado como vehículo en algunas formulaciones. Altas concentraciones de este compuesto se han asociado con toxicidad y necesidad de interrumpir el tratamiento, por lo que en caso de estar disponible es preferible el uso de formulaciones desprovistas de propilenglicol. La SR es el único tratamiento posible en aquellos casos no controlados con tratamiento médico o que desarrollan toxicidad, siempre que la expectativa de vida sea suficientemente prolongada. La SR está indicada también cuando se desea evitar la exposición prolongada a los inhibidores de la esteroidogénesis y en caso de tumores ocultos cuando las expectativas de localización del tumor son escasas34.
El pronóstico del SCE viene determinado por el tipo de tumor responsable, la severidad del hipercortisolismo12,16,24, y la rapidez y eficacia con las que este se controla3,5,12. Asimismo, el pronóstico es más favorable en aquellos casos con tumor oculto que en casos con una fuente de ACTH evidente. En nuestra serie, tras un seguimiento medio de 40 meses, solo 2 pacientes se encontraban en RC, una tasa similar a las reportadas previamente10. Tres pacientes fallecieron en un intervalo de tiempo relativamente corto tras el diagnóstico (media de 19,5 meses), todos ellos con un tumor cuya estirpe es reconocida como de mal pronóstico y con hipercortisolismo severo y de difícil control.
En conclusión, en nuestro estudio encontramos que el SCE es una entidad infrecuente, que se asocia a un amplio espectro de TNE de diferente agresividad y naturaleza. Habitualmente el origen ectópico del SC puede sospecharse por los datos clínicos y bioquímicos y confirmarse en la mayoría de los casos sin necesidad de realizar pruebas invasivas. Su presencia es un dato de mal pronóstico, ya que generalmente se diagnostica cuando el tumor se encuentra en estadios avanzados.
Tanto para el control del hipercortisolismo como del tumor fue necesario el uso de múltiples modalidades terapéuticas. El tratamiento antitumoral resultó en la curación o estabilización de la enfermedad neoplásica en más de la mitad los pacientes, y fue posible el control del hipercortisolismo tras la aplicación de tratamiento médico y/o quirúrgico en todos los casos menos en uno. Dada la complejidad de esta entidad, recomendamos la participación de personal experimentado en el campo de la Oncología y de la Endocrinología para lograr un adecuado manejo, y mejorar su pronóstico.
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.
