




La detección de una neoplasia suprarrenal en el estudio de una hipertensión arterial (HTA) abre un amplio abanico de posibilidades diagnósticas, potencialmente causantes de la propia HTA o independientes de esta. Aportamos un caso con esta asociación, con una peculiar trayectoria diagnóstica y un excepcional diagnóstico final.
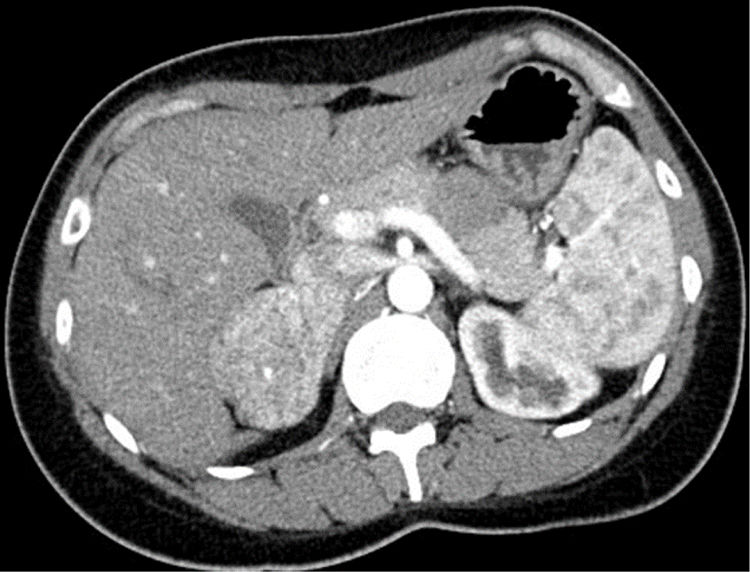
Mujer de 29 años remitida a la consulta de Nefrología tras la detección de HTA por cefaleas de un año de evolución. Intervenida a los 5 años de estenosis pieloureteral izquierda, refería polaquiuria reciente con nicturia. Aportaba un registro ambulatorio con presión arterial (PA) media diurna y nocturna de 154/100 y 140/90mmHg. En la analítica de su médico de atención primaria destacaba una caliemia baja en dos determinaciones (3,49 y 3,12mEq/l), con función renal normal. El estudio ampliado en el servicio de Nefrología evidenció una actividad de renina plasmática baja (0,2ng/ml/h) con aldosterona normal-baja (7,3ng/dl), catecolaminas urinarias normales y un angioTAC con una masa suprarrenal derecha hipervascular de 6cm de diámetro (fig. 1). Consultado el servicio de Endocrinología se indicó ampliar el estudio con metanefrinas urinarias fraccionadas, 3-metoxitiramina urinaria y cortisol libre urinario, todos ellos normales, además de ACTH (34,5pg/ml) y DHEAs plasmáticas (1,31μg/ml), junto con la determinación en suero de cromogranina A y testosterona, todos ellos normales. Evaluada en consulta con estos resultados, no manifestaba alteraciones menstruales ni hirsutismo, negaba el consumo habitual de regaliz y su fenotipo era normal. Se indicó cirugía preferente por el tamaño de la masa tumoral, y en el suero almacenado en la seroteca hospitalaria se solicitó simultáneamente la determinación de metabolitos intermedios de la esteroidogénesis en laboratorio de referencia. Intervenida una semana después mediante laparotomía subcostal ante la potencial malignidad, se resecó una suprarrenal derecha de 87,1g que contenía una tumoración no encapsulada sólida homogénea amarillenta de 6,5cm, con un fino ribete de tejido adrenal comprimido. El estudio anatomopatológico mostró un crecimiento tumoral de patrón trabecular y tubular-pseudoglandular sin mitosis, necrosis, calcificación ni invasión vascular. La inmunohistoquímica mostró positividad para Melan-A, inhibina y CKA1-AE3, con negatividad para cromogranina, S100, CEA, EMA y renal cell marker; todo ello compatible con adenoma suprarrenal, con Ki67 <2%. Tras la intervención se recibieron los resultados de los análisis pendientes, con niveles de desoxicorticosterona (DOCA) casi 16 veces por encima del límite superior de la normalidad (LSN) (237,9ng/dl; LSN: 15), con 11-desoxicortisol y corticosterona normales. Evaluada a los 15 días se había normalizado la PA, desaparecido la poliuria y la caliemia era normal (4,32mEq/l). Al mes de la cirugía, la bioquímica sérica mostraba valores normales de DOCA (7,1ng/ml), con niveles aún bajos de actividad de renina. Pasados 2 años y medio de la intervención se mantiene asintomática, con PA normal y sin lesiones en el lecho quirúrgico en una RM abdominal.

La posibilidad de una forma secundaria de HTA debe plantearse ante la presencia de algunos síntomas clínicos, principalmente cefalea, taquicardia o hipersudoración que apunten un exceso de catecolaminas, o de alteraciones analíticas, en particular la hipocaliemia espontánea1. Esta alteración electrolítica, en ausencia de una pérdida digestiva o iatrógena por diuréticos, sugiere la posibilidad de un exceso mineralocorticoide. Dentro de este grupo el aldosteronismo primario es la causa más frecuente2, y se caracteriza por la secreción autónoma de aldosterona en un adenoma unilateral o por ambas glándulas en la hiperplasia bilateral. Las demás variantes, incluida la hiperplasia unilateral, los adenomas múltiples, los carcinomas productores de aldosterona (aislada o cosecretada con otros esteroides) y las formas genéticas, son mucho menos frecuentes. En todas estas presentaciones, la renina, o bien medida por actividad, o bien como masa, estará baja o suprimida, con niveles normales o altos de aldosterona (usualmente por encima de 15ng/dl), no suprimibles con la sobrecarga salina. Otra forma de exceso mineralocorticoide aparente se produce con el hipercortisolismo severo por rebosamiento del receptor glucocorticoide y escape de la inactivación por la 11ß-hidroxiesteroide deshidrogenasa tipo 2 (11ß-HSD2), habitualmente asociado a producción ectópica maligna de ACTH y sin fenotipo Cushing por la explosividad de su curso3. El consumo excesivo de regaliz, a través del bloqueo del mismo enzima inactivador, origina que el cortisol en niveles fisiológicos active el receptor mineralocorticoide, con la consecuente supresión de la renina, retención salina e hipercaliuria4. Este mismo mecanismo, con base genética, subyace en el denominado síndrome de exceso aparente de mineralocorticoide, debido a mutaciones heterocigotas del enzima 11ß-HSD25. El exceso no tumoral de otros precursores de la esteroidogénesis como causa de exceso mineralocorticoide se puede detectar en edad pediátrica asociado a déficits enzimáticos congénitos, en particular el de 11ß-hidroxilasa6, con exceso de 11-desoxicortisol y DOCA (ambos con efecto mineralocorticoide), y en defectos en la síntesis de andrógenos (17α-hidroxilasa y 17,20-liasa), con alteraciones gonadales asociadas7. El exceso de precursores de la esteroidogénesis asociado a procesos neoplásicos sin síndrome de Cushing es excepcional, dependiente del nivel e intensidad del bloqueo enzimático en el seno de tejido tumoral, y es valorable por estudios de cromatografía que demuestran el perfil de secreción en cada tumor. Habitualmente se describe asociado a carcinomas adrenales8, y excepcionalmente a adenomas suprarrenales, unilateral como este caso9, o bilaterales10. La paciente aportada reúne una serie de características muy inusuales, en especial la sorprendente detección de un tumor adrenal productor de DOCA aislada, y en particular de una neoplasia benigna pese a su tamaño y apariencia radiológica. La diferencia entre un adenoma y un carcinoma suprarrenal depende de criterios no del todo precisos antes de la intervención, y por ello una disección quirúrgica cuidadosa en los casos con potencial malignidad es fundamental. La reversión clínica, bioquímica y radiológica tras la intervención confirma el origen tumoral de la DOCA, sin ningún otro metabolito esteroidogénico elevado, indicativo de una producción selectiva de esta hormona en el seno de un adenoma radiológicamente agresivo por su tamaño y vascularización.
En conclusión, se aporta un nuevo caso de un tumor suprarrenal productor de DOCA, con la peculiaridad de su benignidad, curso favorable y ausencia de hipersecreción de otros metabolitos esteroideos.
AutoríaMiguel Paja: concepción, diseño y escritura de manuscrito. Adela L. Martínez: recogida de datos y su interpretación. Andoni Monzón: recogida de datos y su interpretación. Javier Espiga: atención y seguimiento clínico del paciente; revisión crítica del artículo.
A la paciente por su colaboración para esta publicación.
