




A 42-year-old male with no relevant family or personal history attended the emergency room complaining of severe headache, nausea, and vomiting, and was found to have blood pressure of 220/100mmHg and hyperglycemia. His brain CT scan was normal. Later, he experienced pain in his right hypochondrium. Abdominal ultrasonography showed a right retroperitoneal mass, and a thoracoabdominal CT scan confirmed a right adrenal lesion (16×13×15cm) suggesting adrenal carcinoma, plus a conglomerate of mediastinal lymph nodes.
The patient was admitted to the urology department, and, his laboratory profile was completed in collaboration with Endocrinology: normal hemoglobin, PTH, calcitonin, basal cortisol, ACTH, DHEAS, and androstenedione; elevated transaminases, enolase 226ng/mL (0–16.3), chromogranin A 1004ng/mL (<450); metanephrines 4748.85μg/24h (20–374), normetanephrines 80,233.96μg/24h (30–778), and vanillylmandelic acid 10.1mg/24h (1–7.3) in 24h urine.
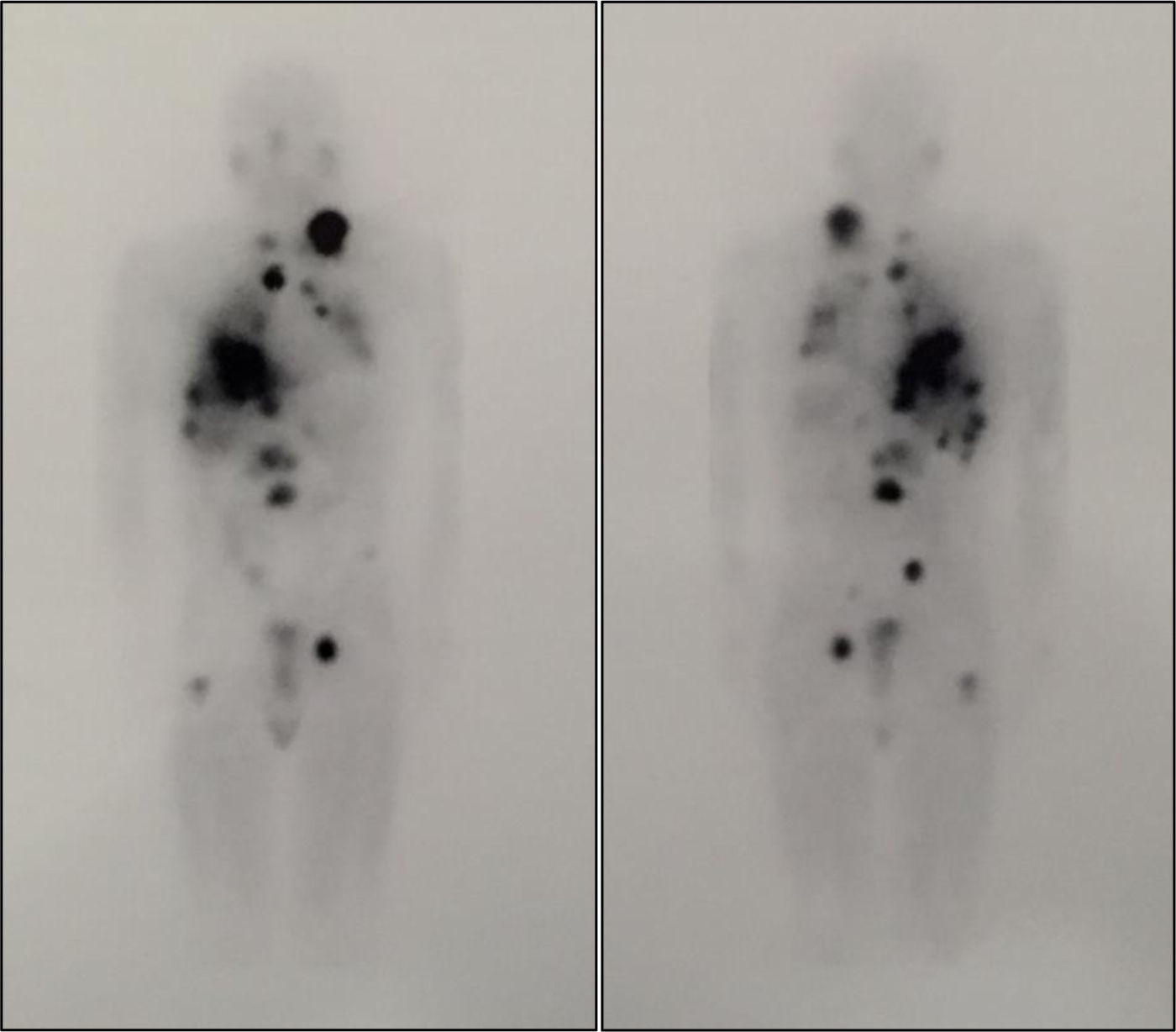
Scintigraphy and SPECT-CT with metaiodobenzylguanidine (131I-MIBG) revealed an increased uptake in the right adrenal gland and disseminated nodal (cervical mediastinal and interaortocaval lymph nodes), liver, and bone. A differential diagnosis between pheochromocytoma metastasis and multiple paragangliomas was considered.
A biopsy of an interaortocaval lymph node showing a Zellballen pattern with large basophilic granular cytoplasm and extensive capsular and vascular infiltration was reported as pheochromocytoma metastasis.1
Due to elevated normetanephrine and metanephrine levels and the value of genetic information as a guide to management and targeted treatment, SDHx,1,2 VHL, and RET genetic testing was conducted, which was negative.2–5
The patient underwent surgery 27 days after alpha-adrenergic blockade with doxazosin (4mg/24h) and seven days after beta-adrenergic blockage with propranolol (10mg/8h) due to sinus tachycardia.1,6 Right adrenalectomy and nephrectomy were performed, and a liver nodule was resected for histological examination. Surgery was hemorrhagic, with an initial trend to high blood pressure, and subsequent hypotension refractory to vascular filling. Metanephrine (2472.5μg/24h) and normetanephrines (23,775.6μg/24h) levels decreased in the early postoperative period.
The histology of the adrenal mass was consistent with pheochromocytoma; there was infiltration of the adrenal capsule, the vasculature and periadrenal adipose tissue, with necrosis and pleomorphism, and an aggressive behavior. The resected liver nodule was metastatic pheochromocytoma (this finding in non-chromaffin tissue is a criterion of malignancy).5 A pathological examination of the adrenal gland and liver showed a mitotic index <1mitosis/10 HPF, Ki-67 of 3%, and positive immunohistochemical staining for synaptophysin and chromogranin.
The disease progressed, and 4 months later, debulking surgery of metastases with resection of a mediastinal mass (19cm) and a nodule in the upper lobe of the left lung (1.5cm) was performed. A subsequent thoracoabdominal CT scan confirmed the progression of the disease. It showed a left supraclavicular adenopathic conglomerate, left paraaortic mediastinal adenopathies, lung nodules, liver metastases, a large interaortocaval mass, and a heterogeneous nodule (3cm) in the adrenalectomy bed.
Two fractionated doses of 100mCi of 131I-MIBG7 were administered with a two- month interval, and were well tolerated. A whole body scan after the first dose revealed disease progression, with lesions with high radiotracer affinity (Fig. 1). By contrast, after the second dose, uptake decreased in some lesions, and there was a slight size reduction, especially in the lungs, with no new adenopathies or distant metastases. Metanephrine and normetanephrine levels markedly decreased three months after the second dose (metanephrines: 677.29μg/24h, normetanephrines: 11,215.51μg/24h), with no changes in lesion size seen in the CT scan.
.")
Whole body scan after treatment with 100mCi of 131I-MIBG. The image shows disease progression with lesions with high affinity for the radiotracer that correspond to a left laterocervical adenopathic conglomerate, mediastinal adenopathies, and lung, liver, and bone metastases (L2 and proximal third of right femur).
Six months later, in view of the persistence of the disease, treatment was started with systemic chemotherapy (cyclophosphamide-vincristine-dacarbazine),7 but had to be discontinued due to toxicity (vomiting, kidney failure, and pancytopenia). Finally, sunitinib (a tyrosine kinase inhibitor) was prescribed at a dose of 50mg/day, but discontinued after four days due to clinical worsening.7 The patient died after a catecholaminergic crisis in the setting of sporadic stage IV malignant pheochromocytoma. His overall survival was 17 months.
Up to 30% of pheochromocytomas and paragangliomas originate in germline mutations;2,5 the remaining tumors are sporadic, and 30% of them originate in somatic mutations.4,8 The prevalence of pheochromocytoma malignancy is 10%–15%.1,5
Knowledge of the mutated gene and the type of mutation (germline or somatic) is important for the genetic diagnosis of family members, in determining the best imaging technique for follow-up,5 and when considering therapeutic targets aimed at the abnormal molecular pathway.4
Mutations are classified in two groups based on their transcriptional profile. The first group, associated with the hypoxic response, encompasses the SDH complex and VHL. The second, associated with activation of the kinase cascade and protein translocation, includes the NF1 and RET genes. The catecholamine secretion pattern guides the genetic study; the predominance of dopamine or normetanephrine suggests mutations in SDHx or VHL, while the predominance of metanephrine points to RET. In the reported case, both gene groups were tested because the catecholamine elevation was mixed. Immunohistochemical staining for SDHB after surgery is advised to guide genetic testing, because negative results suggest mutations in the SDHx genes (SDHC, SDHA, SDHAF2) or large deletions in these same genes.5,9,10 However, this technique was not available at our hospital.
The first therapeutic option is surgical debulking after preparatory treatment with alpha- and beta-adrenergic blockade. Subsequently, patients with 123I-MIBG uptake should be prescribed adjuvant therapy with 131I-MIBG. If no response is seen or progression occurs after treatment with radiolabeled drugs, chemotherapy with cyclophosphamide, vincristine and dacarbazine is recommended.5
The molecular therapies being developed combine temozolomide-thalidomide, sunitinib, or somatostatin analogs. Other options under study, targeted to the SDHB mutation, the most aggressive and frequent in malignant pheochromocytoma (metastasis in 40%),1,2,5 include 2-oxoglutarate and ascorbate.4,8,9 However, the estimated five-year survival rate in malignant pheochromocytoma is less than 50%.6,7
Please cite this article as: Campos-Fernández J, Aguillo-Gutiérrez E, Agudo-Tabuenca A, Medrano-Navarro AL, Borau-Maorad L. Dolor abdominal y crisis hipertensiva como manifestación inicial de un feocromocitoma maligno. Endocrinol Nutr. 2017;64:178–180.
