







Clinically non-functioning pituitary adenomas (NFPAs) are among the most common tumors in the sellar region. These lesions do not cause a hormonal hypersecretion syndrome, and are therefore found incidentally (particularly microadenomas) or diagnosed based on compressive symptoms such as headache and visual field defects, as well as clinical signs of pituitary hormone deficiencies. Immunohistochemically, more than 45% of these adenomas stain for gonadotropins or their subunits and are therefore called gonadotropinomas, while 30% of them show no immunostaining for any hormone and are known as null cell adenomas. The diagnostic approach to NFPAs should include visual field examination, an assessment of the integrity of all anterior pituitary hormone systems, and magnetic resonance imaging of the sellar region to define tumor size and extension. The treatment of choice is transsphenoidal resection of the adenoma, which in many instances cannot be completely accomplished. The recurrence rate after surgery may be up to 30%. Persistent or recurrent adenomas are usually treated with radiation therapy. In a small proportion of these cases, drug treatment with dopamine agonists and, to a lesser extent, somatostatin analogs may achieve reduction or at least stabilization of the tumor.
Los adenomas hipofisarios clínicamente no funcionantes son los tumores más frecuentes de la región selar. Dado que estas lesiones no resultan en un síndrome de hipersecreción hormonal, se manifiestan por síntomas compresivos como cefalea y alteraciones campimétricas, así como por manifestaciones clínicas de hipopituitarismo, o bien son descubiertos de forma incidental (en particular los microadenomas). Inmunohistoquímicamente, más del 45% de estos adenomas inmunotiñen para gonadotropinas o sus subunidades, por lo que se los conoce como gonadotropinomas; mientras que el 30% de los casos no inmunotiñe para ninguna hormona y se los denomina adenomas de células nulas. El abordaje diagnóstico de los adenomas hipofisarios clínicamente no funcionantes debe incluir la evaluación de los campos visuales y la medición de las hormonas de la hipófisis anterior, así como una resonancia magnética nuclear para establecer el tamaño y la extensión del tumor. El tratamiento de elección es la resección transesfenoidal del adenoma, que en ocasiones no se logra completamente. La tasa de recurrencia después de la cirugía puede ser de hasta el 30%. Los adenomas persistentes o recurrentes suelen ser tratados con radioterapia. Una proproción pequeña de estos pacientes puede responder de forma favorable a agonistas dopaminérgicos y, en menor medida, a análogos de la somatostatina.
According to the most recent report of the Central Brain Tumor Registry of the United States (CBTRUS), 15.5% of all central nervous system (CNS) neoplasms are pituitary tumors, third only to meningiomas (37.1%) and glioblastomas (15.6%).1 In young adults, aged 20–34 years, over 30% of CNS tumors are in fact pituitary adenomas (PA).1 These figures are similar to those reported by a meta-analysis, whereby the prevalence of pituitary adenomas in autopsy and radiological studies was found to be 16.7% and 22.5%, respectively.2 It most be noted however, that many of these adenomas are small, incidentally found asymptomatic lesions.2–5 The age-adjusted incidence rate of PA is estimated to be 3.4 cases per 100000 inhabitants per year.2
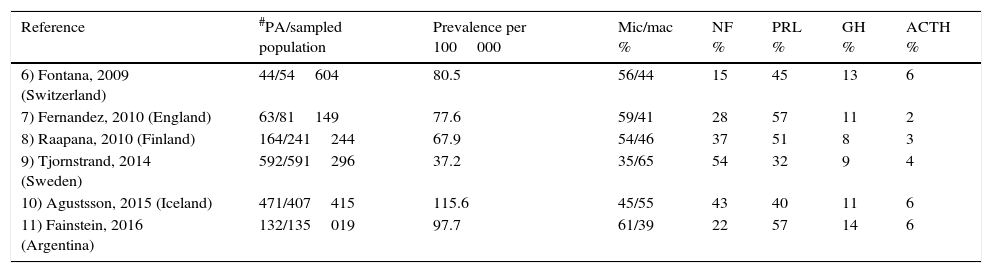
Among community-dwelling studies, clinically non-functioning pituitary adenomas (NFPA) account for a mean of 33% of all PA, ranking second only to prolactinomas that represent 47% (Table 1).6–11 NFPA are the most common type of adenomas when taking into account only macroadenomas, whereas prolactinomas predominate when both micro- and macroadenomas are considered in the analysis. Since they do not result in a hormonal hypersecretion syndrome, the diagnosis of NFPA is either made incidentally or relies on the detection of symptoms and signs of mass effect such as headaches and visual abnormalities due to optic chiasm compression as well as pituitary hormone deficiencies.3,12,13 The prevalence of NFPA varies between 60 and 100 cases per million inhabitants, with a bimodal peak incidence between the ages of 25–45 and 60–70 years and a standardized incidence rate of 1.02–1.08 per 100000; there is no gender predominance.4,9,11,13,14
Prevalence of pituitary adenomas among different community-dwelled studies and the proportion of the different adenoma subtypes.
| Reference | #PA/sampled population | Prevalence per 100000 | Mic/mac % | NF % | PRL % | GH % | ACTH % |
|---|---|---|---|---|---|---|---|
| 6) Fontana, 2009 (Switzerland) | 44/54604 | 80.5 | 56/44 | 15 | 45 | 13 | 6 |
| 7) Fernandez, 2010 (England) | 63/81149 | 77.6 | 59/41 | 28 | 57 | 11 | 2 |
| 8) Raapana, 2010 (Finland) | 164/241244 | 67.9 | 54/46 | 37 | 51 | 8 | 3 |
| 9) Tjornstrand, 2014 (Sweden) | 592/591296 | 37.2 | 35/65 | 54 | 32 | 9 | 4 |
| 10) Agustsson, 2015 (Iceland) | 471/407415 | 115.6 | 45/55 | 43 | 40 | 11 | 6 |
| 11) Fainstein, 2016 (Argentina) | 132/135019 | 97.7 | 61/39 | 22 | 57 | 14 | 6 |
Pituitary adenomas are benign, epithelial neoplasms, arising from a single cell clone that has undergone one or several mutational events.15–17 A small proportion of these tumors, approximately 5%, occur in the context of hereditary syndromes such as type 1 multiple endocrine neoplasia (MEN1) caused by inactivating mutations of the menin gene,18 the Carney complex resulting from inactivating mutations in the regulatory alpha-subunit of protein kinase-A (PRKARA1)19 or the familial isolated pituitary adenoma syndrome (FIPA) due to loss of function mutations of the aryl hydrocarbon receptor binding protein gene (AIP).20 Although in these hereditary syndromes the molecular abnormalities leading to tumor formation have been relatively well characterized, in the vast majority of pituitary adenomas occurring sporadically these molecular changes are only infrequently found as somatic events.21 In fact, NFPA are far less frequent than functioning lesions in these hereditary syndromes. Data from a multicenter European study shows that among 324 cases of MEN1, 136 harbored pituitary adenomas, of which 116 (85.3%) were secreting tumors and only 20 (14.7%) were non-functioning adenomas.22 NFPA have not been reported to occur in the context of the Carney complex, whereby the vast majority of pituitary lesions are GH-secreting adenomas.23 Similarly, the majority of patients belonging to FIPA kindreds harbor GH and to a lesser extent, PRL secreting adenomas; less than 10% have NFPA.24
Thus, the oncogenic mechanisms responsible for the development of sporadic pituitary adenomas in general and of NFPA in particular are likely to involve multiple, simultaneously or sequentially occurring abnormalities in cell cycle regulation.15–17 Some of these abnormalities may take place at an epigenetic level, through the silencing (by hypermethylation of CpG islands) of genes like p16, that encodes a Cyclin D inhibitor that normally prevents a cell with damaged DNA from progressing beyond the G1 phase of the cell cycle.25 The pituitary tumor-transforming gene (PTTG1) localized in the short arm of chromosome 5 encodes a protein known as securin that regulates the separation of sister chromatids and modulates the DNA-repair actions of p53 (26). PTTG1 mRNA is overexpressed in a significant proportion of both, secreting and non-secreting pituitary adenomas27 and correlates with markers of aggressive biological behavior such as the Ki-67 index.28 In a recently published whole-exome sequencing study of 7 patients with NFPA, no MEN1, PRKRA1 or AIP germline mutations could be identified in DNA from peripheral mononuclear cells, and although 24 somatic genetic variants were found in tumoral DNA, including genes such as PDGF (platelet derived growth factor), ZAC (zipper sterile α motif kinase) and NDRG4 (N-myc down regulated gene family member 4), these were not thought to be pathogenically relevant.29
One of the most intriguing aspects of PA biology is that only 0.1–0.2% progress into malignant tumors with metastasis, despite some of them being highly invasive and having considerable recurrence rates.30–33 In fact, this is particularly true for NFPA, since almost 90% of the less than 180 cases of primary pituitary carcinoma reported in the English literature develop in hormone secreting tumors, usually PRL and ACTH, and to a lesser extent GH and TSH.30–33 Although the precise mechanisms responsible for this relative resistance of PA to undergo malignant transformation are poorly defined, the highly specialized, hormone-secreting cells of the anterior pituitary have intrinsic restraints that prevent uncontrolled proliferation and promote senescence and apoptosis.15,16 The hallmarks of pituitary tumor senescence include the induction of cyclin inhibitors such as p16 and p21, which leads to Rb protein hypophosphorilation.34–36 Hypophosphorilated Rb binds the transcription factor E2F, which can no longer promote the progression into the S phase of the cell cycle.34–36 The latter results in chromosomal condensation into distinct heterchromatic foci, aneuploidy and the expression of the lisosomal enzyme β-galactosidase.34–36 There is experimental evidence in PA that oncogenic signals such as PTTG1, which are initially permissive for pituitary tumor formation, subsequently lead to defective DNA replication and aneuploidy.26,37 Studies looking at gene expression profiles at either the mRNA or protein level in pituitary carcinoma have variably found up-regulation of CCND1 (Cyclin D1), VEGF (vascular endothelial growth factor), MMP-9 (Matrix metalloproteinase-9) and several micro RNAs, and down-regulation of MGMT (O6-methylguanine-DNA-methyltransferase), p16ink4A and p27kip1 (Cyclin D1 inhibitors); Bcl-2, Bax and Bcl-X (genes encoding apoptotic proteins); and MT3 (Methalothionein-3).38
Clinical presentationNon-functioning pituitary microadenomas are diagnosed incidentally when an imaging study, be it MRI or CT is performed for unrelated reasons.3,4,12 Although usually benign, non-functioning pituitary macroadenomas constitute a heterogeneous group of tumors with varying biological behaviors that range from clinically asymptomatic lesions to rapidly growing neoplasms that compromise the integrity of the optic chiasm and cranial nerves.39 The clinical manifestations of non-functioning pituitary macroadenomas are the result of the mass effect of the tumor.4,12,13,39 A throbbing headache and visual field abnormalities are among the most commonly reported complaints occurring in 60–80% of the patients, respectively.4,12,39,40 Although visual field defects (usually bitemporal hemianopia) are common,41,42 the prevalence of oculo-motor abnormalities due to involvement of cranial nerves III, IV and VI is very low (less than 5%) even in patients whose tumors invade the cavernous sinuses.4,12,13 In fact, the presence of oculo-motor abnormalities occurring in the context of a large pituitary mass should prompt the suspicion of alternative diagnosis such as infiltrative disorders like sarcoidosis43,44 or histiocytosis45 as well as pituitary metastasis from other primary tumors.46,47
Approximately 8–10% of patients with NFPA presents with pituitary apoplexy.48 Pituitary apoplexy is an acute clinical syndrome characterized by the sudden development of headache, vomiting, altered consciousness, visual abnormalities and hemodynamic instability in the context of hemorrhagic infarction of a pituitary adenoma.49–52 It occurs in 5–20% of all pituitary tumors and appears to be more common in NFPA than in hormonally active lesions.50,52 Although pituitary apoplexy may develop spontaneously, several potential precipitating causes have been described and include dynamic testing with hypothalamic releasing hormones, treatment with dopamine agonists and the concurrent treatment with anticoagulant and antiplatelet drugs.50,52,53
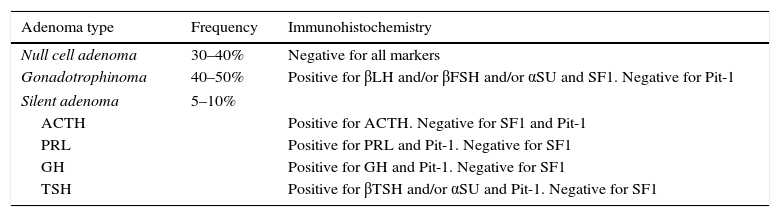
PathologyPA had been traditionally classified according to their staining characteristics with hematoxylin and eosin (HE) as acidophilic, basophilic and chromophobic.54 In general, PRL and GH-secreting adenomas are acidophilic (or eosinophilic), whereas ACTH adenomas are basophilic and NFPA are chromophobic upon HE staining.54 With the emergence of ever more sophisticated immunohistochemistry (IHC) techniques, we can determine the precise cellular lineage of the adenoma using specific antibodies against the hormones that the cells produce.55,56 More than 45% of NFPA show positive immunostaining for α-subunit as well as for LHβ and/or FSHβ and therefore are in fact gonadotrope cell adenomas or gonadotrophinomas (Table 2).55–57 Consistent with their cellular lineage, gonadotrophinomas immunostain positively for steroidogenic factor-1 (SF-1) and are negative for Pit-1, which are both transcription factors crucial in pituitary cytodifferentiation (Table 2).55,58 In approximately 30–40% of cases, NFPA do not immunostain for any hormone at all and are known as null cell adenomas (Table 2).55,59 A small proportion of NFPA, perhaps not more than 10%, immunostain for ACTH, GH, PRL or TSH and are thus termed silent corticotrope, somatotrope, lactotrope or thyrotrope adenomas, respectively (Table 2).55,60–63 Not infrequently, these silent adenomas immunostain for more than one hormone, and are known as plurihormonal adenomas.64 According to some authors silent adenomas can have a more aggressive biological and clinical behavior than gonadotrophinomas or null cell adenomas.55,60–63
Immunohistochemical phenotypification of clinically nonfunctioning pituitary adenomas.
| Adenoma type | Frequency | Immunohistochemistry |
|---|---|---|
| Null cell adenoma | 30–40% | Negative for all markers |
| Gonadotrophinoma | 40–50% | Positive for βLH and/or βFSH and/or αSU and SF1. Negative for Pit-1 |
| Silent adenoma | 5–10% | |
| ACTH | Positive for ACTH. Negative for SF1 and Pit-1 | |
| PRL | Positive for PRL and Pit-1. Negative for SF1 | |
| GH | Positive for GH and Pit-1. Negative for SF1 | |
| TSH | Positive for βTSH and/or αSU and Pit-1. Negative for SF1 | |
SF1: steroidogenic factor 1.
NFPA express somatostatinergic and/or dopaminergic receptors in varying proportions.65–67 The proliferative index is established by immunostaining the tumor sample with a monoclonal antibody against a nuclear protein known as Ki-67, which is expressed only in proliferating cells.68 The Ki-67 index is usually expressed as the percentage of nuclei that positively immunostain with the antibody, and while it can be as high as 50% in very aggressive tumors such as glioblastoma multiforme it is usually 1–3% in PA, especially silent corticotrope adenomas.65,69,70
The term atypical pituitary adenoma was coined in 2004 by an expert panel of the World Health Organization to describe a subset of pituitary tumors, that albeit not fulfilling the diagnostic criteria for carcinoma (namely, the presence of distant metastasis) displayed an aggressive biological behavior.71,72 Histopathologically, these atypical adenomas are characterized by a high mitotic activity with Ki-67 indexes greater than 3%, excessive p53 immunoreactivity and distinctive morphological features such as large pleomorphic nuclei and prominent nucleoli.71,72 Virtually all atypical adenomas are large macroadenomas invading either the cavernous or sphenoid sinuses and/or with suprasellar extension.73 NFPA account for half of these atypical tumors, the majority of them being null-cell adenomas.73
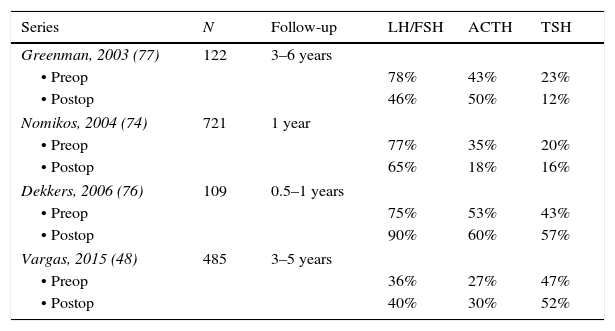
Hormonal abnormalitiesMost patients with NFPA have evidence of one or more anterior pituitary hormone deficiencies.40,48,74–77 The abnormalities in pituitary function are due to both, the interference with the secretion of hypothalamic releasing hormones and the destruction of anterior pituitary cells.4,12,13 The most frequently found hormonal deficiency is hyposomatotropinemia in 70–80% of cases, followed by hypogonadotropic hypogonadism in 40–75% of cases; central hypothyroidism and hypocortisolism are somewhat less frequent, occurring in approximately 20–40% of the patients (Table 3).40,48,74–77 Over 20% of patients with NFPA have two or more hormonal deficiencies.40,48,74–77 Vasopressin deficiency is rare at diagnosis although postoperative diabetes insipidus can be variably found in 15–30% of patients undergoing transsphenoidal surgery (TSS) and 5–10% of these patients end up with permanent diabetes insipidus.40,48,74
Prevalence of pituitary hormone deficiencies at diagnosis and after pituitary surgery.
| Series | N | Follow-up | LH/FSH | ACTH | TSH |
|---|---|---|---|---|---|
| Greenman, 2003 (77) | 122 | 3–6 years | |||
| • Preop | 78% | 43% | 23% | ||
| • Postop | 46% | 50% | 12% | ||
| Nomikos, 2004 (74) | 721 | 1 year | |||
| • Preop | 77% | 35% | 20% | ||
| • Postop | 65% | 18% | 16% | ||
| Dekkers, 2006 (76) | 109 | 0.5–1 years | |||
| • Preop | 75% | 53% | 43% | ||
| • Postop | 90% | 60% | 57% | ||
| Vargas, 2015 (48) | 485 | 3–5 years | |||
| • Preop | 36% | 27% | 47% | ||
| • Postop | 40% | 30% | 52% | ||
Mild elevations in serum PRL, usually below 100ng/mL, can be found in one third of patients with NFPA.40,78,79 The hyperprolactinemia in these patients results from the interruption of the descending dopaminergic tone due to stalk compression by the adenoma.78 It is of paramount importance to differentiate this scenario from a PRL-secreting adenoma, because while the treatment of choice for NFPA is TSS, dopamine agonists constitute the mainstay of treatment for prolactinomas. In this regard, although macroprolactinomas usually result in prolactin levels greater than 1000ng/mL, in some patients the saturation of the primary antibody of the immunoassay by massive amounts of analyte may lead to spuriously low PRL concentrations, the so called “hook effect”, which can be avoided by diluting the serum sample prior to the assay.80
Although over 45% of NFPA are in fact gonadotrope cell adenomas, they seldom produce a hormonal hypersecretion syndrome.12,13,65,81 Nevertheless, in some patients certain patterns of the LH and FSH serum concentrations and their subunits can make us suspect that we are in fact dealing with a gonadotrophinoma.62,82,83 In men, the isolated elevation of FSH in the presence of a low LH and testosterone is diagnostic of an FSH secreting gonadotrophinoma.84 Also in men, the rare scenario of a marked elevation of LH and testosterone, regardless of the serum FSH level and usually accompanied by testicular hyperplasia are indicative of a LH-secreting gonadotrophinoma.84 In normal postmenopausal women with an intact gonadotrophic axis both, serum LH and FSH are concordantly elevated along with a low or undetectable estradiol level; finding a disproportionately elevated LH is highly indicative of a gonadotrophinoma.85 In both pre and postmenopausal women with hypogonadotrophic hypogonadism LH and FSH are concordantly diminished and accompanied by a low estradiol concentration.62,85 In premenopausal women, the hypersecretion of FSH can give rise to ovarian hyperstimulation, which is characterized by amenorrhea/oligomenorrhea associated with giant ovarian cysts.86 In both men and women with gonadotrophinomas stimulation with exogenous thyrotropin releasing hormone (TRH) results in a characteristic rise in LH β-subunit.84,85
In most cases, the diagnosis of central hypothyroidism can be established by the finding of a low free thyroxine concentration in the presence of a low or inappropriately normal TSH and there is no need for dynamic testing.87 Similarly, a morning cortisol concentration below 5μg/dL along with a normal or reduced ACTH is diagnostic of central hypocortisolism.87 When the morning cortisol is between 5 and 15μg/dL we recommend performing an insulin-induced hypoglycemia test; a rise in serum cortisol to 20μg/dL or more is indicative of an intact hypothalamic–pituitary–adrenal axis (HPA).87 When the insulin-induced hypoglycemia test is contraindicated because of the concomitant presence of a seizure disorder or heart failure an alternative way of assessing the HPA axis is the synthetic ACTH stimulation test.87 Although the gold standard for the diagnosis of GH deficiency in the adult patient is the GH response to insulin induced hypoglycemia,87 the finding of an IGF-1 concentration below the lower limit of normal for age, confidently establishes the diagnosis, particularly in the setting of two other pituitary hormone deficiencies.88
Imaging studiesMRI of the sellar region is the ideal method to evaluate pituitary adenomas and it should include both coronal and sagittal sections of less than 3mm.89,90 In T1-weighted images, the adenomas can be hypo- or isointense compared to non-tumoral pituitary tissue and take up gadolinium poorly or not at all (Fig. 1A).89,90 In T2-weighted images the adenomas appear isointense compared to the white matter (Fig. 1B).89,90 When there is bleeding into the tumor, as in cases of pituitary apoplexy the hemorrhage appears as hyperintense in the T1-weighted images without contrast.89,90 Other non-adenomatous lesions of the sellar region like meningiomas or craniopharingiomas are more heterogeneous and usually take up gadolinium more avidly.89,90 Nishioka et al. retrospectively correlated the histological subtypes of 390 NFPA with preoperative MRI findings.91 These authors found that MRI findings such as giant lesions (>4cm), marked cavernous sinus invasion and a lobulated configuration of the supraselar tumor were significantly more common among silent ACTH, GH, TSH and PRL adenomas than among null cell or gonadotrope adenomas.91
Natural history of NFPA
Studies that have evaluated this issue consist mostly of retrospective series with a limited number of patients with follow up periods that go from months to over 10 years and in which the rate of tumor growth or progression varies between 20% and 50%.92–98 Fernandez–Balsells systematically analyzed some of these studies looking for tumor growth progression and other adverse outcomes in untreated patients with NFPA.99 The overall incidence of tumor growth per 100-person-years was 5.75 (95% CI, 4.59–6.51) and it was considerably higher in patients with macroadenomas (12.53, 95% CI 7.86–17.20) than in those with microadenomas (3.32, 95% CI 2.13–4.5) and in patients with solid lesions (5.72, 95%CI 2.28–9.16) compared to those with cystic lesions (0.05, 95% CI 0–0.18).99
Pituitary surgeryThe decision to treat or not to treat a patient with NFPA must take into account several factors, some of which pertain the tumor itself and some others that are related to the individual characteristics of each case (Fig. 2). For instance, withholding surgery could be appropriate in asymptomatic patients without visual or hormonal abnormalities, harboring relatively small, incidentally found adenomas located at a reasonable distance from the optic chiasm as long as a close surveillance plan with follow up MRI and visual field examination is implemented; this approach is particularly valid in young subjects who wish to preserve fertility as well as in elderly patients with a high cardiorespiratory anesthetic risk (Fig. 2). At the other end of the spectrum there are patients in whom acute or even emergency surgery is indicated because of progression of tumor growth with impending visual compromise or intracranial hypertension and in cases of pituitary apoplexy with hemodynamic instability (Fig. 3).
 tumor remnant is demonstrated in the postoperative MRI and the patient is asymptomatic, has relative preservation of hormone function and wants to preserve fertility it is reasonable to observe and monitor the lesion periodically. Pituitary surgical reintervention could be attempted when there is an accessible tumor remnant. Tumor remnants measuring more than 10mm and particularly when located in the cavernous sinuses should undergo radiation therapy. Choosing between conventional external RT or radiosurgery will depend on the size and location of the remnant and its proximity to the optic apparatus. (TSS: transsphenoidal surgery; MRI: magnetic resonance imaging; XRT: external radiotherapy; RadioQx: radiosurgery).")
Management algorithm for nonfunctioning pituitary adenomas at our center. Incidentally found microadenomas and small, intrasellar macroadenomas are managed conservatively. Extrasellar macroadenomas, particularly when symptomatic, are subjected to TSS. If a small (less than 6mm) tumor remnant is demonstrated in the postoperative MRI and the patient is asymptomatic, has relative preservation of hormone function and wants to preserve fertility it is reasonable to observe and monitor the lesion periodically. Pituitary surgical reintervention could be attempted when there is an accessible tumor remnant. Tumor remnants measuring more than 10mm and particularly when located in the cavernous sinuses should undergo radiation therapy. Choosing between conventional external RT or radiosurgery will depend on the size and location of the remnant and its proximity to the optic apparatus. (TSS: transsphenoidal surgery; MRI: magnetic resonance imaging; XRT: external radiotherapy; RadioQx: radiosurgery).

MRI of a patient with a large and invasive clinically nonfunctioning pituitary macroadenoma treated with 2.5mg per week of cabergoline. The patient presented with headaches and bitemporal hemianopia and had serum prolactin levels below 10ng/mL on several occasions; surgical debulking of the lesion was attempted unsuccessfully but the excised specimen revealed a chromophobe adenoma that positively immunostained only for FSH. Note the reduction in tumor volume at 6 and 12 months of treatment.
The treatment of choice of NFPA is the transsphenoidal resection of the tumor. TSS should be performed by an experienced, high-volume pituitary neurosurgeon and it could be accomplished by the endoscopic or microscopic approaches. Both techniques present advantages and limitations and ultimately the decision as to which of the two is used in the individual patient depends on the neurosurgeon's experience and preference.100–102
Although the majority of cases can be operated via the transsphenoidal approach, the transcranial route is indicated when dealing with large tumors extending into the median fossa and the cavernous sinuses.100,101 Whereas the mortality rate for TSS is below 1%, transcranial operations carry a mortality risk of up to 6%.4,13,100,101 Complete removal of the adenoma by TSS can be achieved in 40–90%.48,74–77 Recently, our group reported the clinical characteristics and surgical outcome of 483 patients with NFPA.48 In this series that recapitulates the experience of two pituitary neurosurgeons, a complete resection of the adenoma was achieved in 317 of the 428 patients (74%) who were operated by TSS and in 22 of the 38 (58%) in whom a transcranial approach was used.48 The two most significant factors associated with recurrence were the presence of extension into the cavernous sinus and of an adenoma remnant after initial surgery.48 In concordance with these findings, Reddy et al. reported a recurrence rate of only 6.9% in patients without evidence of a remnant on postoperative MRI, while 40% of the patients with a tumor remnant had tumor re-growth after 5–10 years of follow up (103). While visual abnormalities and other manifestations resulting from the mass effect of the pituitary tumor significantly improve after successful resection of the adenoma, recovery of the pituitary hormone deficiencies is less frequent (Table 3).48,74–77,102,103
Perioperative managementThe perioperative management in our center follows a local protocol derived from over 20 years of experience treating these patients. Patients with known preoperative central hypothyroidism and hypocortisolism should ideally be well replaced by the time they get to surgery. We use stress doses of hydrocortisone even in patients with an intact corticotrophic axis. Our protocol consists of hydrocortisone 100mg IV upon induction of anesthesia, continuing on with 100mg IV every 8h for the first postoperative day and tapering it down to 50mg every 12h by the second postoperative day. Patients with known central hypocortisolism are usually discharged from hospital on either prednisone 5mg daily or oral hydrocortisone 10mg in the morning, 5mg at noon and 5mg in the early evening. Patients with a normal corticotrophic axis preoperatively are sent home without glucocorticoid replacement.
The diagnosis of transient DI is established by a sharp rise in urinary output (usually more than 250mL/h), particularly when associated with a drop in urine osmolality and an elevation of serum sodium. We treat transient DI with subcutaneous desmopressin acetate injections 5–10μg every 8–12h as needed to keep urinary volumes below 150mL/h. If by the second postoperative day the patient is still requiring desmopressin to keep a normal diuresis, the possibility of permanent DI is entertained and the regimen is switched to oral desmopressin 0.1–0.2mg every 12h.
Postoperative follow upEven in expert hands, NFPA have a recurrence rate of approximately 20–30%, therefore a systematic MRI surveillance protocol is mandatory in order to implement adjunctive therapies (Fig. 2).70 The first postoperative MRI is usually obtained 3–6 months after surgery and thereafter on a yearly basis for the first 5 years (Fig. 2).70,103 If no recurrences are found the frequency of imaging studies can be decreased to every 2 years for the next 6 years.70,103 Small tumor remnants or recurrences without visual field or ocular abnormalities may be treated conservatively with observation and yearly MRI, depending on the individual case (Fig. 2).
It is controversial to what extent pituitary hormonal deficits recover after surgery. Some authors describe a postoperative recovery of gonadotrope and thyrotrope function in 12–32% and 4–11% of patients, respectively,74,77 whereas others report no functional improvements or even worsening of these axes48,76 (Table 3). It seems that the ACTH deficiency found in NFPA is the hormonal deficit less likely to recover after pituitary surgery.48,74,76,77 In our center, pituitary reserve is evaluated 1, 3, 6 and 12 months postoperatively in order to ascertain the development of new deficits and to identify patients with recovering axes.
Radiation therapyA significant proportion of these tumors will recur after TSS upon long-term follow up.48,74–77 Unfortunately, there are neither clinical/imaging characteristics, nor histopathological features or biomarkers capable of accurately predicting recurrences and therefore indicate the need for prophylactic therapeutic interventions such as postoperative radiation therapy (XRT). Although there is extensive evidence supporting the effectiveness of postoperative XRT in preventing recurrences, most if not all published studies have been retrospective and suffer from a selection bias whereby XRT is reserved for the largest and more invasive tumors.105,106 Postoperative XRT can reduce the recurrence rate of NFPA from 40% in non-radiated patients to less than 10% in those who are radiated.105,106 However, the benefits of postoperative XRT should be weighed against the potential side effects, including the development of pituitary hormone deficiencies, cerebrovascular events, optic nerve injury, and more rarely, the emergence of secondary brain tumors.105–108 Although hypopituitarism is practically unavoidable following XRT, pituitary hormone deficiencies are highly prevalent in patients with NFPA even at diagnosis and are unlikely to improve after surgery, even in patients not receiving XRT.50,74–76 Radiation-induced optic neuritis occurs in 1–3% of cases and is manifested by an acute loss of vision.104–106 This complication has been attributed to microvascular obliteration of the visual tract and is directly related to the radiation dose and the proximity of the tumor to the optic chiasm; an advanced age and the concomitant presence of diabetes appear to increase the risk.106 The use of modern radiation techniques with carefully planned dosing schedules has made this complication a very infrequent event.106 The development of secondary brain tumors after pituitary radiation is extremely rare and has been estimated to be around 2% at 20 years follow up.107 A reduced life expectancy has been reported in patients with pituitary tumors treated with XRT.108 The increased mortality rate, which in most cases is due to cardio- and cerebrovascular events, has been linked to hypopituitarism and is better documented in patients with GH-secreting adenomas than in patients with NFPA.108 XRT has also been linked to a reduced quality of life in some but not all studies.109–111
In view of the available data, withholding XRT in patients with a complete adenoma resection and no evidence of tumor remnants on postoperative MRI seems undoubtedly reasonable, particularly if there are few or no anterior pituitary hormone deficiencies (Fig. 2).
Pharmacological treatmentSince NFPA frequently express variable proportions and subtypes of somatostatinergic (SSTR) and dopaminergic receptors (D2R), the pharmacological treatment of these tumors with somatostatin analogs and/or dopamine agonists constitutes an attractive alternative.65–67 Using a standardized in vitro experimental protocol Florio et al. evaluated the expression of SSTR subtypes and D2R as well as the inhibition of [3H]-thymidine incorporation by octreotide, cabergoline and the chimeric compound BIM23A760 (both, a somatostatin analog and a dopamine agonist) in 38 primarily cultured NFPA (31 gonadotrophinomas and 7 null cell adenomas).112 While D2R mRNA was expressed in all tumors, SSTR2 and SSTR3 mRNA were found at somewhat lower levels in 80% of the tumors, whereas only 4 tumors expressed SSTR5 mRNA.112 A 22–28% inhibition of [3H]-thymidine incorporation was observed in 48%, 66%, 54% and 63% of the tumors treated with octreotide, cabergoline, a combination of octreotide and cabergoline and the chimeric compound BIM23A760, respectively.112
Unfortunately, the evaluation of the efficacy of such pharmacological therapy is limited by the lack of a measurable biochemical marker, in contrast to what occurs in functioning adenomas. In NFPA the only indicator of a therapeutic response is the reduction in tumor size documented by long term, serial imaging studies. The very limited clinical data available to date indicates that somatostatin analogs achieve a significant reduction in tumor size in only 12% of patients.113,114 The experience with dopamine agonists like cabergoline is somewhat more optimistic because 30–50% of treated patients show a significant reduction in tumor volume (greater than 25%) after 6–12 months of treatment (Fig. 2).115 In a recent multicenter study Greenman et al. retrospectively evaluated the outcome of CBG treatment of 79 patients with NFPA who had residual tumor remnants after TSS; in 55, CBG was administered preventively, whereas in 24 it was indicated after the remnant showed evidence of regrowth.115 One third of the patients treated preventively showed evidence of tumor volume reduction, whereas in 49% the tumor remained stable and in 13% an increase in size was detected. Stabilization of the tumor mass was achieved in 53% of the patients who received CBG after adenoma regrowth.115
ConclusionDespite being among the most common lesions of the sellar region, no formal consensus guidelines for the diagnosis and treatment of NFPA have been developed by any of the world endocrinological or neurosurgical organizations. In the last two decades we have witnessed striking advances in the pathogenesis, diagnosis and treatment of pituitary diseases that have led to a significant improvement in the quality of life and a sharp reduction in standardized mortality rates of patients suffering from conditions like acromegaly, Cushing's disease and prolactinoma.116 While such advances favorably impact patients with NFPA as well, an individual with a non-secreting pituitary macroadenoma often will require several therapeutic interventions in order to prevent or manage recurrences, including pituitary surgical re-interventions and/or the use of radiotherapy in any of its modalities (Fig. 2). In contrast to functioning adenomas for which several effective and relatively safe targeted pharmacological therapies have been developed, a specific medical treatment for NFPA is still lacking.
Conflict of interestThe authors declare that there is no conflict of interest regarding the publication of this paper.
