




The risk of suffering from gonadal germ cell tumors (GCT) is increased in some patients with different sexual development (DSD), mainly in those with Y chromosome material. This risk, however, varies considerably depending on a multitude of factors that make the decision for prophylactic gonadectomy extremely difficult. In order to make informed recommendations on the convenience of this procedure in cases where there is potential for malignancy, this consensus guide evaluates the latest clinical evidence, which is generally low, and updates the existing knowledge in this field.
El riesgo de padecer tumores gonadales de células germinales (TCG) se encuentra incrementado en algunos pacientes con desarrollo sexual diferente (DSD), fundamentalmente en aquellos que presentan material de cromosoma Y. Dicho riesgo, sin embargo, varía considerablemente en función de multitud de factores que dificultan enormemente la decisión de una gonadectomía profiláctica. Con el fin de hacer recomendaciones fundamentadas sobre la conveniencia de este procedimiento en los casos donde existe potencial de malignización, esta guía de consenso evalúa la última evidencia clínica existente, en general escasa, y actualiza los conocimientos en este terreno.
A 2006 consensus statement established the term "disorders of sex development" (or, more commonly used now, "differences of sex development" [DSDs]), and reorganised diagnoses into three broad groups based primarily on sex chromosomes and secondarily on hormonal aetiology and genetics.1
Conditions considered DSDs have wide-ranging aetiologies and phenotypic expressions; however, just some of them in particular, especially those with Y chromosome material, feature a predisposition towards developing neoplasms. These are, essentially, gonadal germ cell tumours (GGCTs) (≃14.9% of all DSDs; 0.8%–40% depending on age and diagnosis) and, with a lower prevalence (0.9%) and only in some DSD subtypes, gonadal stromal tumours and smooth-muscle leiomyomas/hamartomas.2,3
With advances in molecular diagnostics technology, numbers of identified DSD-causing genes have expanded considerably, allowing for a more accurate taxonomy. Nevertheless, determining the personalised risk of gonadal neoplasia (essentially a GGCT) in a person with DSD remains a daunting task, considering that said risk varies considerably depending on multiple other non-genetic factors.
In order to make substantiated recommendations on the advisability of prophylactic gonadectomy in cases in which the potential for malignant transformation is non-negligible, this article offers an update on knowledge in this regard and evaluates the latest existing clinical evidence, which is generally limited, on this potential in each group or condition included among DSDs.
Everything set out below will exclusively refer to the risk of GGCTs, since these represent the most common strains of gonadal malignancies in DSD patients, and since histopathological tests can predict the risk of suffering from them.
Germ cell ontogenyGerm cell development is a very complex process strictly organised in time and space on a genetic basis that is not entirely well understood. It starts in week two with somatic cells that, through a process of specification driven by BLIMP1 and SOX17, transform into primordial germ cells (PGCs). Guided by KIT/KITLG signalling, they migrate in groups along the midline from the proximal epiblast towards the gonadal ridge; this process occurs between weeks five and six, and with it, they become gonocytes (testicles) or oogonia (ovaries) and remain morphologically indistinguishable from PGCs. During this period, epigenetic reprogramming takes place which grants them the ability to transfer the characteristic of pluripotentiality to the next generation. This phenomenon is due to factors including genes of parental origin subjected to genomic imprinting involving expression of placental alkaline phosphatase (PLAP), c-KIT, octamer-binding transcription factor 3/4 (OCT3/4) and other markers.4–6
Once the undifferentiated gonad (gonadal ridge and germ cells) is formed, initiation of a signalling route around week seven will determine the differentiation thereof as a testicle or an ovary. In particular, the SRY and SOX9 genes will be the main drivers of testicular differentiation, working through multiple transcription factors such as NR5A1 and ZFPM2 to form Sertoli cells, while the development of the ovary will require the absence of SRY expression at the same time as initiation of WNT4/β-catenin, FOXL2 and RSPO1, leading to differentiation of stromal cells in granulosa cells. In the testicle, due to interaction with pre-Sertoli cells, gonocytes will differentiate into pre-spermatogonia which, as part of their maturation process, will be displaced from the center to the basal lamina of the seminifeous tubule, losing expression of embryonic markers OCT3/4 and c-KIT to then mature into spermatogonia and acquire expression of DDX4 and TSPY. With all this, testicular differentiation will end in week nine, when the Leydig cells are capable of producing testosterone (T) and insulin-like factor 3 (INSL3) and the Sertoli cells are capable of producing anti-Müllerian hormone (AMH). Full ovarian differentiation, for its part, will end around week 11 with theca and granulosa cells.
Ultimately, the presence of testicular hormones (T and AMH) will determine male (internal and external) genital differentiation, and the absence thereof will determine female (internal and external) genital differentiation.5,7
Pathogenesis of precursor lesions to germ cell tumours in differences of sex developmentBy definition, primordial germ cells are the most pluripotent cells in the body following embryogenesis, in which hypomethylation and gene expression are similar to those of embryonic stem cells. In most patients with DSDs, the germ cells disappear as the body grows by means of a process of apoptosis. Some may persist in a state of immaturity; these are generally believed to be the cells that are vulnerable to malignant transformation, becoming a pre-invasive lesion or a precursor to an invasive GGCT.
DSD patients at risk of GGCTs are those who, in the presence of germ cells, have Y chromosome material in their gonadal karyotype; this occurs regardless of whether or not any degree of testicular differentiation is present. This Y chromosome material includes certain genes involved in male gonadal differentiation found in the region around the centromere of the Y chromosome (GBY region): TSPY (a gene that encodes "testis-specific protein on Y chromosome", located in Yp11.2) and other candidate genes including SRY and DYZ3.8
Starting from these premises, the problem seems to lie in the fact that, in the absence of duly developed Sertoli cells, the primordial germ cells that migrate to the gonad in development retain their foetal phenotype. Under these conditions of biological immaturity, germ cells can undergo malignant transformation by the action of the above-mentioned genes of the GBY region, heightened expression of which promotes cell proliferation, as well as prolonged expression of OCT3/4.2,4,6–12 Another essential requirement is for any abnormalities that may occur over the course of gonadal development to have a sufficient impact on the location or environment of these germ cells, but not ultimately affect their survival; otherwise, the risk of GGCTs will be considered null if said environment is hostile enough to induce their death.4,12
It can be concluded, then, that the oncogenic risk of these germ cells in the presence of gonadal Y chromosome material depends more on supporting cells (Sertoli cells) and on the microenvironment in which germ cells themselves are found — or, amounting to the same, that said risk is linked to degree of gonadal differentiation, such that the greater the gonadal immaturity, the greater the oncogenesis risk.
Considering all this, it has been estimated that the risk of such abnormalities being maintained after birth and resulting in a precursor lesion to infiltrating GGCT (whether or not it ultimately progresses to an infiltrating GGCT) is 14.9% on average in DSD patients with Y chromosome material. This essentially occurs in patients in whom the underlying genetic defect leads to early blockage of gonadal development (e.g. mutation or deletions of SRY or WT1). This risk is also present, though somewhat lower, in 45X0/46XY mosaicism (gonadal karyotype), while it is significantly lower in DSDs that do not interfere with normal gonadal development but do affect germ cell maturation (e.g. abnormalities in androgen action).2,13
To these intrinsic risk factors should be added others such as the possible role of gonadotropins (especially follicle-stimulating hormone [FSH]), oestrogens and, above all, androgens, which account for the higher risk of oncogenic development at two specific points in life: minipuberty and, in particular, puberty/youth (reinitiation of previously arrested mitosis). Patient age (older age being associated with higher risk) and gonadal location (intra-abdominal gonads carrying a higher risk) are also risk factors.2,7,14
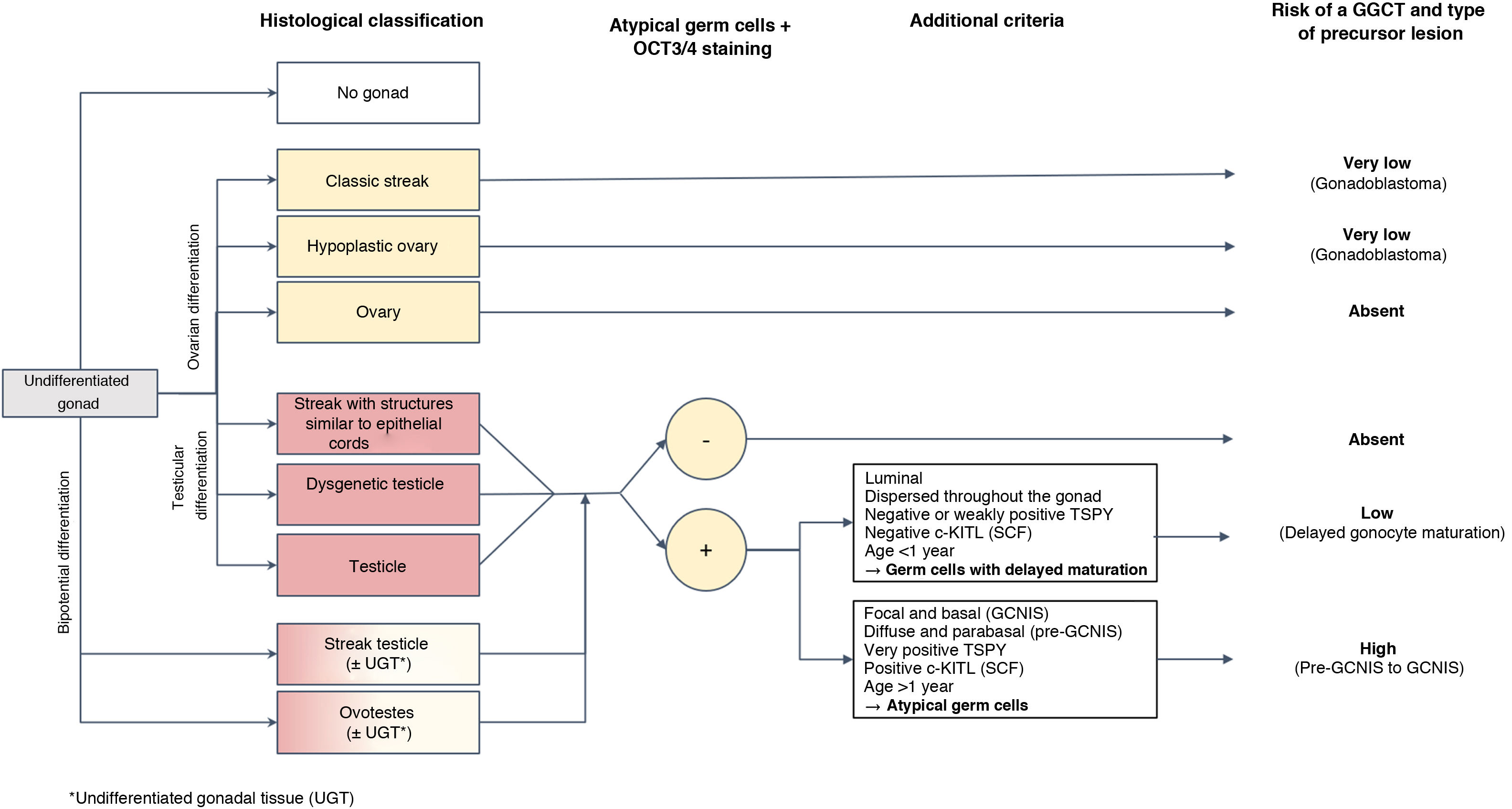
Histopathological diagnosis of precursor lesions in patients with differences of sex development and course of the risk of germ cell tumour based on theseHistological study of the gonads in DSD patients enables classification of the degree of development and maturity thereof, grouping them as follows: (1) classic streak and hypoplastic ovary in gonads with ovarian differentiation; (2) streak with epithelial cords, dysgenetic testicle and structurally normal testicle in gonads with progressive differentiation towards a testicle; and, finally, (3) streak testicle and ovotestis in gonads with bipotential differentiation (Fig. 1).15 This degree of development plus some of the above-mentioned circumstances represent the factors that can contribute in certain DSD patients to their gonads developing precursor lesions (less common in fully developed gonads such as the testicle, ovary or ovotestis) and that, ultimately, can progress late or early to infiltrating GGCT. These precursor lesions are usually generically called germ cell neoplasia in situ/gonadoblastoma (GCNIS/GB). Their diagnosis is based on certain combined histopathological findings (morphology), for cases of DSD featuring testicular tissue and expression of certain immunohistochemistry markers of pluripotentiality.

As mentioned, germ cells in initial stages of ontogenesis express certain immunohistochemistry markers such as OCT3/4 encoded by POU5F1; stem cell factor (SCF), also known as c-KIT ligand (encoded by KITLG); and PLAP. After birth, cells with sustained “delayed maturation,” essentially those of testicular origin, tend to retain positivity for these markers of pluripotentiality until they enter apoptosis. This explains why it is advisable to not rely on them exclusively in histological diagnosis of a precursor lesion to a GGCT of testicular origin, at least in the first 6–12 months of life, and thus prevent overdiagnosis during the postnatal period. To this end, it is crucial to take into account other differentiating histological criteria particular to such lesions such as the distribution of OCT3/4-positive cells in the gonad, the location of these germ cells in the seminiferous tubule, cellular atypia and expression of KITLG. In particular, germ cells with delayed maturation do not express the latter marker (c-KITLG or SCF), tend to be located in the central and suprabasal segment of the seminiferous tubules and are diffusely distributed throughout the gonad, whereas the cells of precursor lesions to GGCTs (GCNIS) express such a factor, will have characteristic atypia, will be in a basal location within the seminiferous tubules in the so-called spermatogonial niche and will have a patchy distribution.5,13
To be precise, dysgenetic gonads that include testicular parenchyma (dysgenetic testicles, streak testicles, streaks with epithelial cords and ovotestes) have a higher incidence of a delayed maturation of germ cells, rarely persisting beyond the first year of life.5,13,16 Therefore, this distinction not exclusively based on the OCT3/4 marker is of paramount importance to prevent erroneous identification as precursor lesions to immature cells in dysgenetic gonads which in the past accounted for incorrect rates of risk of malignant transformation and, therefore, progression to GGCTs.6,7,10,13,17
In the case of dysgenetic gonads featuring Y chromosome material but not testicular tissue (classic streak or streak with epithelial cords), morphology is usually sufficient for diagnosis of a precursor lesion, and the use of immunohistochemistry techniques is not necessary; furthermore, the above-mentioned problem of delayed maturation would not be raised.
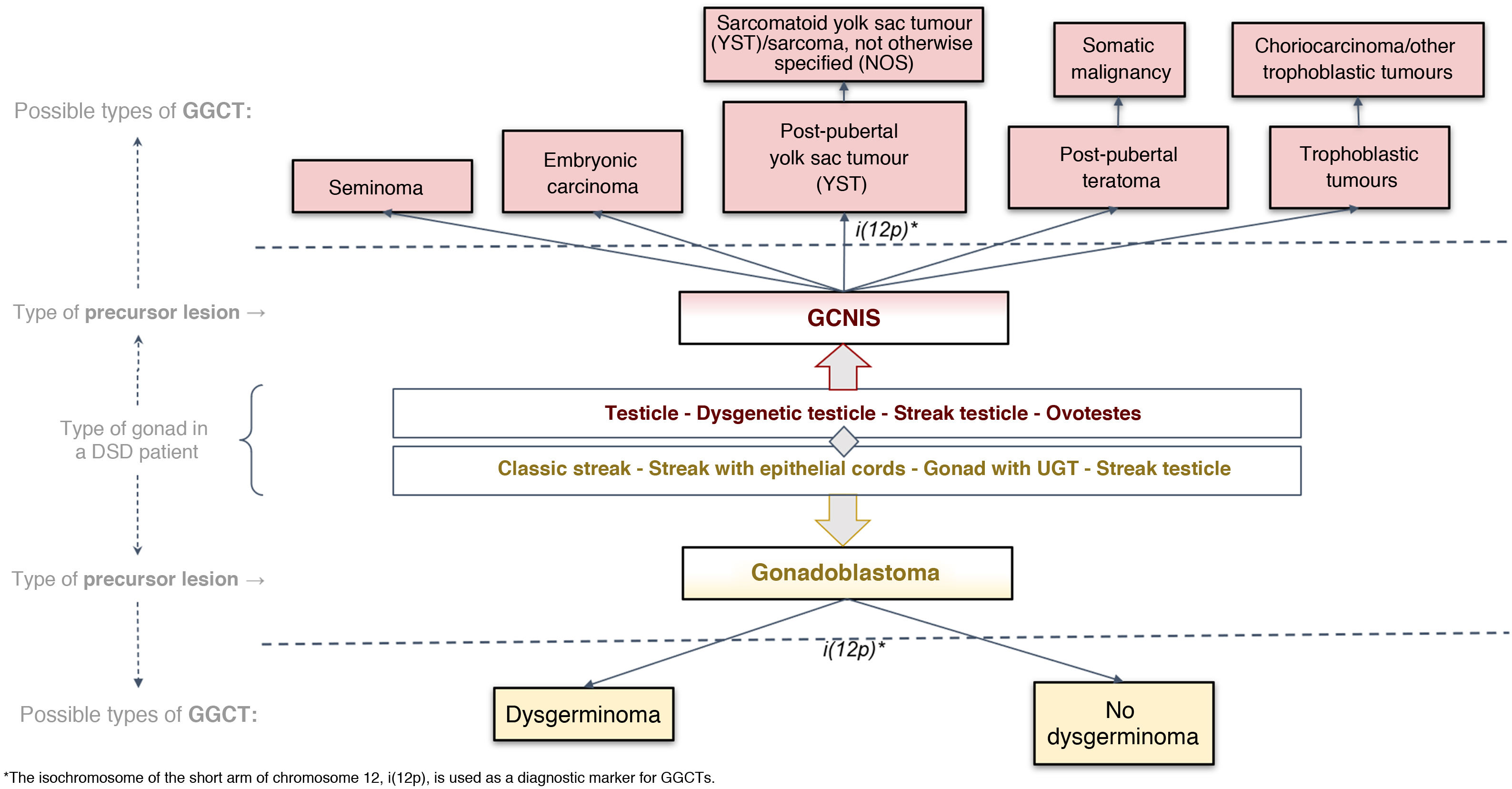
Histological classification of precursor lesions to germ cell neoplasia in situ/gonadoblastoma (GCNIS/GB) and risk of progression to a germ cell tumourBased on histopathological study of the gonadal biopsy specimen and, if applicable, the immunohistochemistry markers mentioned, a diagnosis of a precursor lesion to a GGCT can be made (Fig. 1). These lesions, generically called GCNIS/GB, are subdivided into two types depending on their supporting cells (Sertoli cells or granulosa cells)4,5,7,9–11,13,18–22:
Gonadoblastoma (GB): this usually occurs in DSD patients with streak gonads or hypoplastic ovary; rarely, it occurs in testicular tissue. These patients have an XY karyotype (mosaic or not) but do not express the SRY gene, which would explain why, in many cases, they are phenotypically women. Its development is specifically linked to the GBY, region, which includes the above-mentioned TSPY gene. Thus, 45X0/46XY patients and women with Turner syndrome in the presence of Y chromosome material are at higher risk of GB. Its supporting cells are considered granulosa cells (positive FOXL2) and it is classified in the gropus of non-invasive germ cell neoplasias. Its diagnosis is primarily morphological, and immunohistochemistry is usually not required for diagnosis. It consists of circumscribed nests comprising a mixture of germ cells in different degrees of maturation from very similar to GCNIS (positive for OCT3/4) to similar to spermatogonia and sex cord cells, sometimes surrounded by hyaline deposits of basal membrane material composed of laminin.
Progression from a GB to an infiltrating GGCT is impossible to predict. The actual incidence of this course is unknown given the trend in past decades towards performing prophylactic gonadectomy in these patients, although it has been reported that it may be present at birth or develop later on in life. Some series have found that just 50% become an infiltrating malignant tumour, primarily dysgerminoma, over the course of a patient's life.9,23
GCNIS, as it is called in the latest (2016) WHO classification24; in prior medical literature, other terms were used such as carcinoma in situ (CIS) (which is incorrect as they are not epithelial cells) and intratubular germ cell neoplasia, unclassified type (IGCNU), (which is erroneous in relation to the meaning of unclassifiable). This lesion is considered a precursor lesion to infiltrating GGCT of testicular origin which is located in the seminiferous tubules and which, therefore, takes place in more or less virilised XY patients, due to the presence of the SRY gene. The supporting cells are Sertoli cells (positive for SOX9). GCNIS cells are large and atypical, morphologically similar to a gonocyte/primordial germ cell and found in a basal location in the seminiferous tubule, with extensive clear cytoplasm and a hyperchromatic angulated nucleus with a prominent nucleolus. Its diagnosis may require, in addition to this morphology, immunohistochemistry markers (OCT3/4, c-KIT and PLAP).
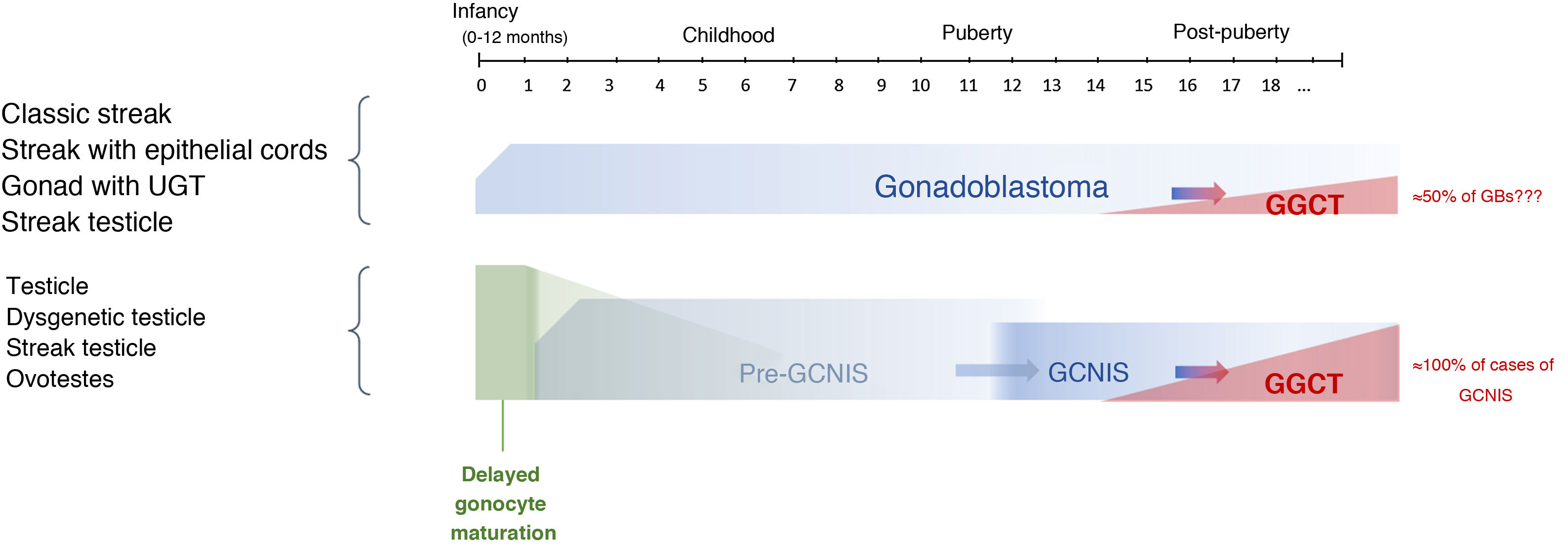
The potential for developing GCNIS is already present at birth as this is a foetal defect in gonocyte maturation, and nearly 100% of cases are predestined (some authors have placed this figure at 70%) to develop as a malignant tumour (GGCT) with a mean age of onset of 14–44 years of age (its onset being most common between age 20 and age 35).9,10,13,23
The term pre-GCNIS or childhood GCNIS was recently proposed to describe an intermediate lesion between delayed maturation and GCNIS that, though suggested as a precursor to the latter, does not always progress to it.25
When one of these precursor lesions (GCNIS or GB) progresses to an invasive tumour, it progresses only to a type 2 GGCT pathways (Fig. 2). According to the latest (2016) WHO classification,23 two pathways include, in the testicle with GCNIS, seminoma and non-seminomatous tumours (embryonic carcinoma; yolk sac tumor, post-pubertal type; teratoma, post-pubertal type; and trophoblastic tumours, the most common of which is choriocarcinoma); and, in streak gonads or ovaries with GB, dysgerminoma (equivalent to testicular seminoma).9,13,23

Histological classification of GGCTs according to the 2016 WHO classification24 applied to dysgenetic gonads.
DSDs: differences of sex development; GCNIS: germ cell neoplasia in situ; GGCTs: gonadal germ cell tumours; UGT: undifferentiated gonadal tissue.
The timeline of this course from initial gonadal tissue to precursor lesions to GCNIS/GB and ultimately to a GGCT is shown in Figure 3.
The risk/benefit balance of prophylactic gonadectomy and decision-making in patients with differences of sex development at risk of germ cell tumours and neoplastic risks (GGCTs) in DSD gonads with Y chromosome material. GCNIS: germ cell neoplasia in situ; GGCTs: gonadal germ cell tumours; UGT: undifferentiated gonadal tissue.")
Prophylactic gonadectomy can only be proposed in DSD patients at risk of GGCTs. The risk of suffering from other neoplastic lineage such as gonadal stromal tumours (referring to Sertoli–Leydig hamartomas, Sertoli cell adenomas and smooth-muscle leiomyomas/hamartomas, characteristic of partial androgen sensitivity syndrome), does not in itself justify prophylactic gonadectomy given that they are uncommon and nearly always benign.
Classically, in decision-making in relation to performing gonadectomy in patients at true risk of GGCTs, three factors have to be taken into account: underlying diagnosis, sex and age at presentation, and molecular histopathological and immunohistochemistry findings (the latter having been recently added).3,8,19 Despite the latest advances in these fields, the above-mentioned risks of malignant transformation (essentially in GBs) amount to mere speculation, as their actual natural history is unknown, in terms of both rates of malignant transformation and times to develop them.4,5,7,17 These limitations mandate consideration for the fact that gonadectomy means infertility, a need for hormone supplementation and a subjective experience reported by some patients of poorer acquisition of secondary sex characteristics, as well as a worsening in quality of life despite suitable replacement therapy. On the other hand, if it is not pursued, despite the fact that the survival rate of a malignant GGCT is high (95% at five years), chemotherapy for these entails lifelong side effects such as a risk of metabolic syndrome and cardiovascular risk.5
That said, today, in DSD patients with gonadal activity (usually testicles), there is a growing trend towards postponing gonadectomy or even avoiding it altogether, carefully balancing the risk of developing a GGCT on the one hand and maintenance of endocrine function and fertility potential on the other hand. In this endeavour, gonadal biopsy remains the gold standard for evaluating this risk in certain DSDs (Table 1), and as a general action plan, it is recommended that the biopsy sample be 3 mm × 3 mm × 2 mm in order to have enough tissue for pathology examination; at the same time, pathologists with experience in this area are needed to ensure suitable interpretation of the lesions. It cannot be overlooked, however, that biopsy does not necessarily represent the entire gonad and that diagnosis of a precursor lesion can be missed; therefore, in gonads with a heterogeneous macroscopic appearance, such as ovotestes, samples should be taken from different areas. In addition, some authors have recommended that biopsy be combined with other procedures such as orchidopexy or sperm extraction in the case of a testicular DSD.7,8,10
Indications for gonadal biopsy in DSD patients.a
|
DSDs: differences of sex development; GCNIS/GB: germ cell neoplasia in situ/gonadoblastoma; GGCTs: gonadal germ cell tumours.
The goal of biopsy is histopathological study and classification of the gonad and, in the case of testicular tissue, depending on morphology, consideration of immunohistochemistry testing that detects a delayed gonocyte maturation (0–12 months of age) versus pre-GCNIS (as of one year of age and before puberty) or GCNIS (usually as of puberty).
Similarly, with the exception of 45X0/46XX or 45X0 Turner syndrome with no Y chromosome material (demonstrated by means of a conventional karyotype, fluorescence in situ hybridisation [FISH] and/or other molecular techniques), if the gonads are suspected to be streaks when they are rudimentary, they should always undergo histological examination to rule out the presence of undifferentiated gonadal tissue (UGT) (a precursor lesion to GB), which has a high likelihood of developing a GGCT over time. For this reason, since from a functional point of view these tissues do not have any sort of hormonal activity, they should always then be removed.10
Depending on the results of the histological examination, before a decision is made as to whether to perform gonadectomy (Table 2), the patient's risks of developing a GGCT should be stratified. In addition, other factors, some of which have already been mentioned, should be considered; these include the patient's chromosome set, underlying diagnosis, age (the older the age, the higher the risk), race (higher risk in Caucasians), anatomical location of the gonad (higher risk in an intra-abdominal location), genital phenotype, potential fertility, quality of gonadal endocrine activity, potential for performing self-examination, risks inherent to surgery and possible side effects of hormone replacement therapy.3,8,10,14 To these factors must be added other, no less important factors of a psychological nature, such as the patient's degree of understanding of risk and cooperation, as well as congruence with the patient's gender identity. All these things must considered together, on the fundamental basis of an evaluation of the degree of development of this identity, when establishing strategies for action and monitoring. Therefore, from the start of the medical approach to a DSD, and essentially during adolescence, the gender identity of these patients should be examined and worked on so that all surgical decisions may be made with the greatest assurances of success.26 Bear in mind, however, that recent studies have shown that most adult individuals with DSDs identify with the gender assigned at birth, at rates close to those of the general population, although when such congruence is not present, rates and degrees of gender dysphoria have been reported as very high.27
Criteria rendering gonadectomy advisable in DSD patients.
|
|
DSDs: differences of sex development; GCNIS/GB: germ cell neoplasia in situ/gonadoblastoma.
When a patient with a DSD, or the patient's parents, ultimately wish to keep the gonads despite the genotype and/or histopathological study and immunohistochemistry testing showing a non-negligible potential for malignant transformation, they can be offered a strategy based on early, proactive detection of localised lesions suggestive of GGCTs through inguinal or scrotal ultrasound imaging and/or through self-examination of the gonads if they are easily palpable (in a scrotal location). If, by contrast, the gonads have an intra-abdominal location, given that imaging has proven insufficient here, alternatives have been proposed such as relocation thereof to the inguinal canal for better visualisation. Whether the reasons for keeping the gonads are these or others, follow-up thereof is advised as of the start of puberty as set out in Table 3.5,8
Proposed follow-up in DSD patients at risk of GGCTsa from the start of puberty to gonadectomy (or indefinitely, if there is a desire to keep the gonads).
|
beta-hCG: chorionic gonadotropin; DSDs: differences of sex development; GGCTs: gonadal germ cell tumours; LDH: lactate dehydrogenase.
This group can include patients in whom biopsy showed no histopathological risk of GGCTs (GCNIS/GB) but was concluded to be potentially non-representative and in whom, furthermore, there are any risk factors such as the presence of testicular tissue, Y chromosome material or intra-abdominal gonad location.
Very significant increases in beta-hCG (by several thousand units) are associated with the presence of choriocarcinoma, while mild increases (by hundreds to one or two thousand units) are associated with the presence of syncytiotrophoblast cells. High levels of alpha-fetoprotein, for their part, are characteristic of yolk sac tumour. There is no increase in such markers in seminoma (or dysgerminoma in the ovary), embryonic carcinoma or teratoma, post-pubertal type.
Finally, whatever the decision made with respect to gonadectomy, the patient and the patient's parents should be informed as to the patient's prospects for fertility. First of all, it is necessary to assess the risk of transmitting the genetically determined DSD condition to one's descendants. It is also necessary to look for germ cells in a gonadal tissue biopsy. Numbers thereof decrease considerably with age, such that they are absent or very scarce after puberty in most of these patients. This cellularity, however, is highly dependent on the patient's condition, such that it may be absent or limited as of birth in a large proportion of cases of gonadal dysgenesis, or it may be normal or almost normal in the beginning, but gradually decrease and exhibit a predominance of immature forms, in patients with complete androgen insensitivity and in patients with mild defects of androgen synthesis such as 5-alpha reductase deficiency. In any case, although the techniques for preserving germ tissue applicable today are feasible (cryopreservation of precursor tissue in early stages of life or, rarely, of sperm or oocytes in some mild cases of DSD with spontaneous pubertal onset), very strict research protocols are required for techniques of induction of future fertility of immature germ cells (maturation in vitro), which have already been achieved for ovarian germ tissue, but are not currently available for those of testicular origin except experimentally.28,29
Proposed indication for prophylactic gonadectomy in differences of sex developmentIn summary, the current recommendations on prophylactic gonadectomy, listed in Table 4, are based on type of DSD. Gonadal radiation has been proposed as an alternative, but experience is limited.8
Proposed consensus for performing gonadectomy by type of DSD.
| Group | Condition | Risk | Recommendation for gonadectomy and follow-up |
|---|---|---|---|
| DSD with abnormalities in the karyotype | 45X0/46XY (mixed gonadal dysgenesis) | Intermediate to high for GGCTs(15%–35%) | Gonadectomy is recommended, although postponing it can be considered depending on degree of virilisation, gender identity and reproductive wishes (if potential for fertility is confirmed):2,8,30,31
|
| 46XY/46XX (chimerism: streak testicle or ovotestes) | Low or very low for GGCTs(2.6%–3%) | Gonadectomy will be indicated in puberty, only on the gonad (testis or ovary) inconsistent with assigned gender5,8,32–35:
| |
| 45X0/46XX(Turner syndrome) | Nearly null or intermediate for GGCTs (GB) depending on whether or not Y chromosome material is detected(0%–1% and 12%–40%, respectively) | All types of Turner syndrome, essentially non-mosaic 45X0 Turner syndrome, require detection of Y chromosome material in the blood (karyotype/FISH followed by molecular testing of genes of the GBY region such as TSPY, SRY and DYZ3)36–41:
| |
| 47XXY(Klinefelter syndrome) | Null for GGCTs | Gonadectomy is not indicated, and neither biopsy nor specific follow-up in relation to the gonads is required. The risk of GGCTs is reported exclusively extragonadally. There have been rare reports of Leydig cell tumours2 | |
| 47XYY | Null for GGCTs | Gonadectomy is not indicated, and neither biopsy nor specific follow-up in relation to the gonads is required | |
| XX DSD with hypervirilisation | Hypervirilisation of NON-gonadal origin (CAH, virilising tumours, etc.) | Null or nearly null for GGCTs | In general terms, gonadectomy is not indicated, and neither biopsy nor specific follow-up in relation to the gonads is required:
|
| Hypervirilisation of gonadal origin (46XX ovotesticular DSD and 46XX testicular DSD) | In the absence of TSPY (± SRY), possibly low or very low risk (2.6%–3%) of GGCTsIn the presence of TSPY possibly intermediate risk of testicular tissue for GGCTs | Gonadal biopsy is usually necessary for a definitive diagnosis in which, furthermore, a karyotype is suggested to rule out gonadal mosaicism, as well as molecular testing of Y chromosome material to determine risk of GGCTs (at least TSPY and SRY)This 46XX gonadal dysgenesis group usually has a genetic cause such as translocation of the SRY gene to the X chromosome or to an autosome (10%–15% of cases of ovotesticular DSD, or 80% of cases of testicular DSD), or, more occasionally, duplication of SOX9, mutations in RSPO1, SOX10, NR5A1, NR2F2, WNT4 or WT1. Few cases feature underlying 46XX/46XY gonadal mosaicism in which the risk of GGCTs seems to be equally low (see 46XY/46XX entity)5,8,32–35Gonadectomy will be indicated in puberty, only on the gonad (testis or ovary) inconsistent with assigned gender. See prior 46XY/46XX section:
| |
| XY DSD | Complete testicular dysgenesis and partial testicular dysgenesis (complete gonadal dysgenesis [CGD] and partial gonadal dysgenesis [PGD])Ovotesticular XY DSDOvarian XY DSD | Intermediate to high for GGCTs (12%–60%) depending on molecular abnormalitiesLow for GGCTs (2.6%–3%)Not reported. Possibly low | Most cases require gonadal biopsy for a definitive diagnosis. Decision-making concerning gonadectomy in this broad subgroup is complicated since the risk of GGCTs varies widely depending on various factors: (1) the presence of the TSPY gene (Yp.11.2) [detection of SRY solely indicates that there is Y chromosome material; it is not useful as a marker of tumour predisposition] and of the causative gene (when found, even though the risks are not usually well established), as well as (2) the characteristics of the gonad: anatomical position (scrotal, inguinal or abdominal) and stage of maturation (undifferentiated or UGT, ovary, testicle or ovotestes): the more mature the gonad and the lower its location, the lower the tumour risk.5,8,12,13,17,44 For more details, the following is proposed:
|
| Abnormalities in androgen synthesis | Not well known (varies by condition) | Although the risk seems generally low or very low, certain conditions seem to carry a risk of GGCTs. Decision-making concerning gonadectomy will depend, therefore, on molecular diagnosis as well as assigned gender5,8,51,52:
| |
| Androgen insensitivity syndrome | Complete forms (CAIS) with no residual AR receptor activity:Low risk of GGCTs#(variable risk across studies: 1%–3% with a cumulative risk of 3.6% at 25 years of age and 33% at 50 years of age,48 other authors have estimated a risk of 15% after puberty49) | As the risk of malignant transformation in adulthood is largely unknown, decision-making with respect to gonadectomy in CAIS remains enmeshed in a great deal of debate. In any case, at present, the most commonly accepted recommendation is gonadectomy in late puberty since: (1) GGCTs (seminoma) present after puberty and (2) if the gonads are kept until post-puberty then spontaneous pubertal feminisation will occur (due to androgen aromatisation) with no need for exogenous oestrogens, thus achieving suitable optimisation of both breast development and bone mineralisation. In addition, (3) postponing gonadectomy enables the patient to take part in decision-making after being duly informed8,53–56:
| |
| Partial forms (PAIS) and CAIS with residual AR receptor activity:Intermediate to high risk of GGCTs (15%–20%)Minor, if a scrotal location; recent studies have substantially reduced this risk | No studies have performed follow-up in patients with PAIS in whom gonadectomy was postponed; therefore, the predominant approach is early bilateral gonadectomy56
| ||
| Patent Müllerian duct syndrome | Low or very low for GGCTs | Gonadectomy is not indicated, nor is biopsy required, although rare cases of GGCTs reported could render follow-up as of puberty or later advisable (see Table 3)3 |
beta-hCG: chorionic gonadotropin; DSDs: differences of sex development; FSH: follicle-stimulating hormone; GBY: region around the centromere of the Y chromosome; GCNIS/GB: germ cell neoplasia in situ/gonadoblastoma; GGCTs: gonadal germ cell tumours; GnRH: gonadotropin-releasing hormone; LDH: lactate dehydrogenase.
