




Most pheochromocytomas and paragangliomas are sporadic lesions. However, up to 40% of them have a hereditary origin, due to germline mutations in some of the 17 genes which to date are known to cause these tumors.1 Among them, the genes of the different succinate dehydrogenase subunits and the VHL gene are the most commonly affected.2
Fumarate hydratase is an enzyme involved in the Krebs cycle, catalyzing the conversion of fumarate to malate. Inactivating mutations of its encoding gene (FH) lead to increased intracellular levels of fumarate, with activation of the pseudohypoxia pathway and the transcription of different genes involved in angiogenesis and tumour growth.3 Heterozygous germline mutations of the FH gene have been previously associated with hereditary leiomyomatosis and renal cell cancer4 (HLRCC, OMIM # 150800), an autosomal dominant hereditary disease characterized by the development of multiple uterine and cutaneous leiomyomas and renal papillary renal cell carcinoma type 2, an aggressive renal tumour with a poor prognosis. In 2013, however, FH mutations were for the first time identified in some patients with hereditary paraganglioma and pheochromocytoma, with an apparently high predisposition to develop metastatic disease.5–7 To date, none of the cases reported in the literature had combined pheochromocytomas or paragangliomas with HLRCC. The present study describes the first such case.
A 44-year-old woman presented in April 2007. Her history included hysterectomy due to uterine leiomyomas. She also had a family history of maternal uterine myomatosis. The patient consulted because of increasingly frequent episodes of palpitations, headache, facial flushing and dizziness, associated with a mild increase in blood pressure. The hormone study revealed plasma normetanephrine >1000pg/ml (normal value [NV]: <180pg/ml), 24-h urine normetanephrine 8045μg/24h (NV: 88–444μg/24h) and chromogranin A 1074ng/ml (NV: 19.4–98.1ng/ml). The plasma and urine metanephrine levels were normal. The abdominal CAT scan revealed a cystic mass in the lower pole of the left kidney, measuring 13cm×13cm×9.6cm in size, with multiple nodular thickenings of the wall that appeared hyperintense after contrast administration (Bosniak grade III cyst). The ipsilateral adrenal gland presented a mass with an enhanced-uptake nodular margin, thick irregular walls, and a large central cystic zone initially considered to be consistent with pheochromocytoma. Following preoperative preparation with doxazosin for three weeks, and propranolol in the last 10 days, the patient underwent left nephrectomy and adrenalectomy, without complications. The histopathological study confirmed the diagnosis of pheochromocytoma measuring 9.5cm in diameter, with necrotic foci and extensive hyalinization areas. The mitotic index was low, with no nuclear pleomorphism and no vascular invasion. The tumour yielded a score of 2, indicating a low risk of metastasis, according to the pheochromocytoma of the adrenal gland scaled score, at the expense of confluent or comedo-type necrosis, and a score of 2 according to the grading of adrenal and extra-adrenal pheochromocytomas and the relationship to prognosis scale, which corresponds to a well differentiated tumour, though Ki-67 expression could not be determined. The nephrectomy specimen also presented a grade 4 renal cell carcinoma according to the Fuhrman classification, with extensive cystic degeneration and a tubulo-papillary pattern, distributed as multiple nodules in the wall of a cyst measuring 10cm in maximum dimension, extending focally outside the capsule and infiltrating the perirenal adipose tissue. After surgery, the plasma and urine normetanephrine and norepinephrine levels were seen to normalize, and the patient's clinical condition was resolved. Since then she has undergone regular controls with repeated abdominal CAT scans, meta-iodobenzylguanidine SPECT scintigraphy and determinations of normetanephrine and metanephrine in 24-h urine, which have discarded local relapse and metastases.
An initial study involving sequencing of the VHL, SDHB and RET genes proved negative. In November 2015, the genetic study was expanded with an analysis of a panel of genes associated with hereditary pheochromocytoma (VHL, RET, EPAS1/HIF2, HRAS, TMEM127, MAX, SDHA, SDHB, SDHC, SDHAF2, FH, MDH2, EGLN1 and NF1),8 which detected a mutation affecting a canonical splicing site in exon 4 of the FH gene (NM_000143.3:c.555+1G>A). This mutation had previously been reported in a Spanish family with cutaneous and uterine leiomyomatosis,9 and in a case of a series of patients with HLRCC in France,10 but not in patients with pheochromocytomas or paragangliomas.
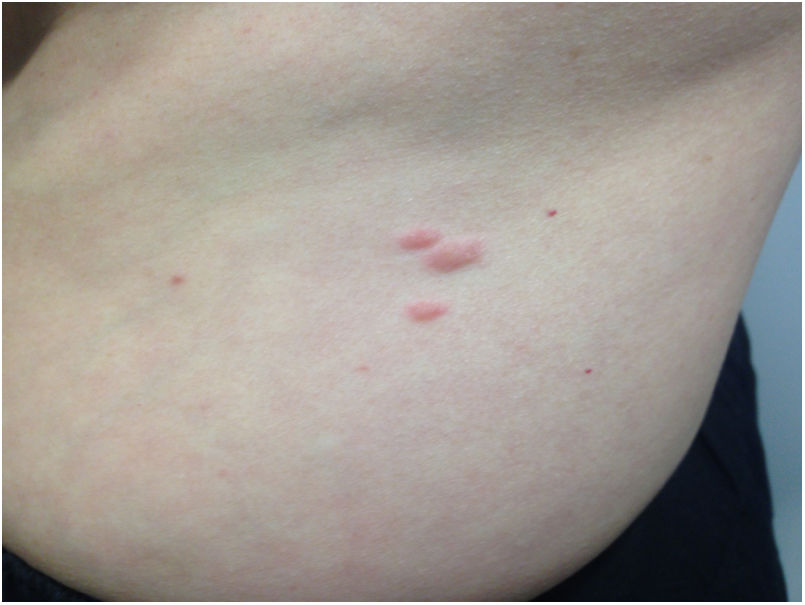
After establishing these results, a more detailed physical examination revealed the presence of pink papules on the forearms and trunk, measuring 0.5–2cm in diameter, and which the patient had noticed for two years (Fig. 1). The biopsy of one of these lesions confirmed skin leiomyoma.

The genetic study of the mutation in the two daughters and a sister of the patient proved negative.
Mutations of the FH gene are responsible for HLRCC syndrome, but are also an exceptional cause of hereditary pheochromocytoma/paraganglioma. To the best of our knowledge, only 7 cases of patients carrying an FH gene mutation with pheochromocytoma and/or paraganglioma have been reported,5–7 three of them with metastatic tumours. None of these patients had cutaneous leiomyomatosis or renal cancer, and only one5 had a history of uterine leiomyomatosis. The present case shows that patients with pathogenic mutations in heterozygosis in the FH gene are at risk of developing both pheochromocytomas/paragangliomas and HLRCC syndrome, and that screening for both disorders should be performed once the genetic diagnosis has been established.
The authors wish to express their gratitude to Dr. Mercedes Robledo (Centro Nacional de Investigaciones Oncológicas, Madrid, Spain), for reviewing the manuscript.
Please cite this article as: Morón M, Afonso JL, Boronat M. Feocromocitoma asociado a leiomiomatosis cutánea y uterina y cáncer renal en un paciente con una mutación germinal en el gen de la fumarato hidratasa. Endocrinol Diabetes Nutr. 2020;67:291–292.
