




Los feocromocitomas son un tipo de tumores neuroendocrinos poco frecuentes que se forman a partir del tejido enterocromafin de la médula adrenal, y pueden sintetizar, acumular y secretar catecolaminas y/o sus metabolitos.
Entre el 3-36% de los paragangliomas y feocromocitomas son malignos. Los asociados a mutaciones de la succinato deshidrogenasa subunidad B (SDHB) tienen mayor riesgo de malignidad.
La enfermedad relacionada con IgG4 (ER-IgG4) es una entidad fibroinflamatoria inmunomediada recientemente descrita, que incluye enfermedades que comparten rasgos anatomopatológicos, serológicos y clínicos. Dentro del espectro de la ER-IgG4 se incluyen entre otras; la tiroiditis de Riedel, la enfermedad de Mikulicz (inflamación de lacrimal, parótida y glándulas submandibulares) o la enfermedad de Ormond (fibrosis retroperitoneal). Se caracteriza por lesiones infiltrativas causadas por un denso infiltrado linfoplasmocitario rico en células plasmáticas IgG4 positivas, que producen un grado variable de fibrosis, con patrón típicamente estoriforme y frecuentemente flebitis obliterante1,2.
Hasta la fecha no se ha comunicado la asociación de feocromocitoma con la ER-IgG4.
Se trata de una paciente de 38 años sin antecedentes personales ni familiares de interés que comenzó en febrero de 2018 con molestias en flanco derecho inicialmente intermitentes y posteriormente continuas, asociadas a anorexia y pérdida de peso. Su médico de familia solicitó una ecografía abdominal que mostró una gran masa redondeada de 10cm de ecoestructura mixta cuyo origen era difícil de precisar, motivo por el que ingresó para estudio. Como hallazgos en la exploración física, además de la masa abdominal palpable, se constataron cifras elevadas de tensión arterial que se trataron con doxazosina requiriendo altas dosis para el adecuado control tensional.
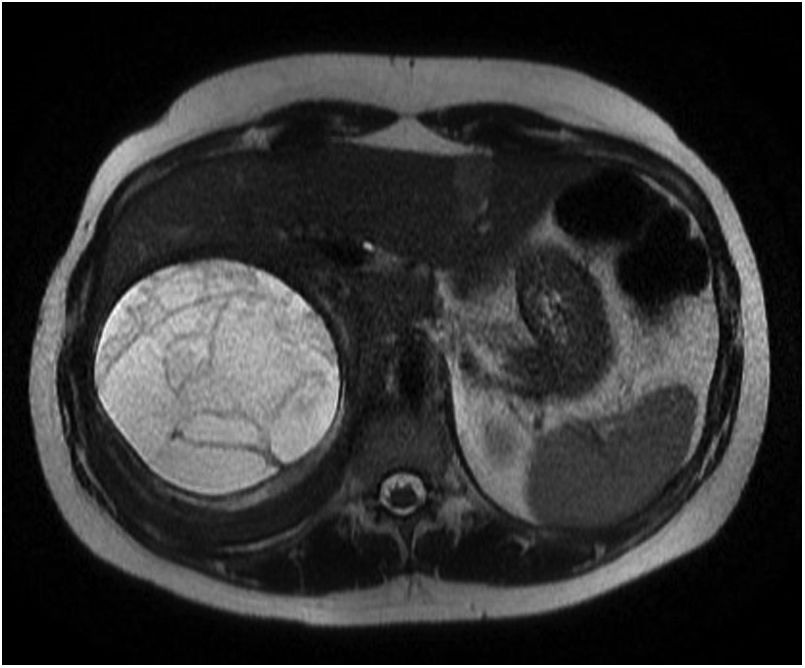
La TAC y la RMN abdominal confirmaron una gran masa adrenal derecha de 11×10×11cm con componente quístico rodeado por una lesión infiltrante retroperitoneal que desplazaba hígado y riñón, y comprimía la vena cava inferior. La lesión en la RMN era hipointensa en las secuencias potenciadas en T2 (fig. 1). Radiológicamente el componente que rodeaba a la parte quística de la lesión tenía aspecto fibroso o granulomatoso.

Se realizó PAAF (sin estudio hormonal previo) de la lesión que descartó malignidad a pesar de las limitaciones que presenta la citología para el diagnóstico.
Posteriormente, se solicitó estudio funcional adrenal que mostró normalidad del perfil del cortisol y andrógenos con elevación de catecolaminas a expensas de normetanefrina más de 4 veces por encima del límite superior de la normalidad, lo que confirmó el diagnóstico de feocromocitoma y se interconsultó a endocrinología.
Se completó el estudio con MIBG más SPECT/TC, que objetivó depósitos dentro de la masa.
En julio de 2018 la paciente se intervino mediante laparotomía abierta practicándose nefrectomía radical con resección de grasa perirrenal y tumorectomía suprarrenal sin incidencias, con buen control tensional desde entonces, normalización de metanefrinas posquirúrgicas, y mejoría de la clínica que presentaba la paciente, aunque persistió durante un tiempo la astenia, anorexia y pérdida de peso no cuantificada de la que ya se ha recuperado.
El estudio anatomopatológico de la lesión adrenal confirmó el diagnóstico de feocromocitoma, y la anatomía patológica de la lesión retroperitoneal fue congruente con la ER-IgG4.
El estudio genético para feocromocitoma fue negativo (se realizó en el Centro Nacional de Investigaciones Oncológicas usando el panel de secuenciación masiva habitual que incluye todos los genes implicados conocidos hasta la fecha). La determinación sérica de IgG4 fue normal. Se completó el estudio mediante PET para conocer el alcance de la enfermedad, que mostró como hallazgo relevante una lesión de partes blandas con intensa captación de FDG, con un SUVmáx de 23,8 que podría corresponder en parte a cambios posquirúrgicos, sin poder descartar recidiva o resto tumoral locorregional. El PET de control, 5 meses después, mostró una disminución tanto del tamaño como de la intensidad de la captación de la lesión, por lo que se interpretó como cambios posquirúrgicos. En la actualidad, la paciente continua en seguimiento con buena evolución clínica y radiológica, a pesar de no haber iniciado tratamiento para la ER-IgG4.
En la ER-IgG4, la elevación sérica de IgG4 solo se produce en aproximadamente el 66% de los pacientes3. La prevalencia exacta es desconocida, en parte por el reciente reconocimiento de la enfermedad.
Esta enfermedad suele afectar a más de un órgano entre el 60-90% de los casos, típicamente órbita, glándulas salivares, páncreas y tejido retroperitoneal, pero también están descritos tiroides, pulmón, pleura, aorta, riñón, ganglios linfáticos, próstata, piel, etc. Se han descrito 4 fenotipos de la enfermedad: enfermedad pancreato-hepato-biliar, aortitis y/o fibrosis retroperitoneal, enfermedad limitada a cabeza y cuello, y síndrome clásico de Mikulicz con afectación sistémica.
La etiología de la enfermedad es desconocida, sin embargo, parece que hay una posible asociación con malignidad, siendo la prevalencia de cáncer hasta 2 veces más frecuente en pacientes afectados por la ER-IgG4 que en la población general. Estudios recientes sugieren que la ER-IgG4 podría ser un signo paraneoplásico de algunos tumores, y por ello habría que tener cautela a la hora de diagnosticar esta reciente enfermedad4,5.
Los pacientes generalmente presentan un desarrollo subagudo de la enfermedad con aparición de una masa en los órganos implicados asociado a molestias relacionadas con la misma, dependiendo de la localización y tamaño. También es frecuente la pérdida de peso hasta que la enfermedad es diagnosticada y tratada.
El tratamiento óptimo de la ER-IgG4 no está bien establecido. Los consensos internacionales sugieren los corticoides como primera línea para inducir remisión en todos los pacientes con enfermedad activa y también en recaídas6.
En nuestro caso nos parece llamativa la presencia de feocromocitoma de características radiológicas atípicas y gran tamaño en una paciente joven que, pese a tener estudio genético completo negativo, sospechamos podría encontrarse en relación con alteraciones genéticas no conocidas. Hasta el momento no hay datos de malignidad, pero dadas las características referidas requerirá un seguimiento estrecho7,8.
Consideramos en nuestro caso excepcional la asociación de 2 entidades muy poco frecuentes como son el feocromocitoma y la ER-IgG4, ya que hasta ahora ambas entidades no se habían descrito conjuntamente. Desconocemos si dicho hallazgo es casual o causal. El mayor conocimiento de la ER-IgG4 en el futuro ayudará a esclarecer la relación real entre ambas enfermedades.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.
