




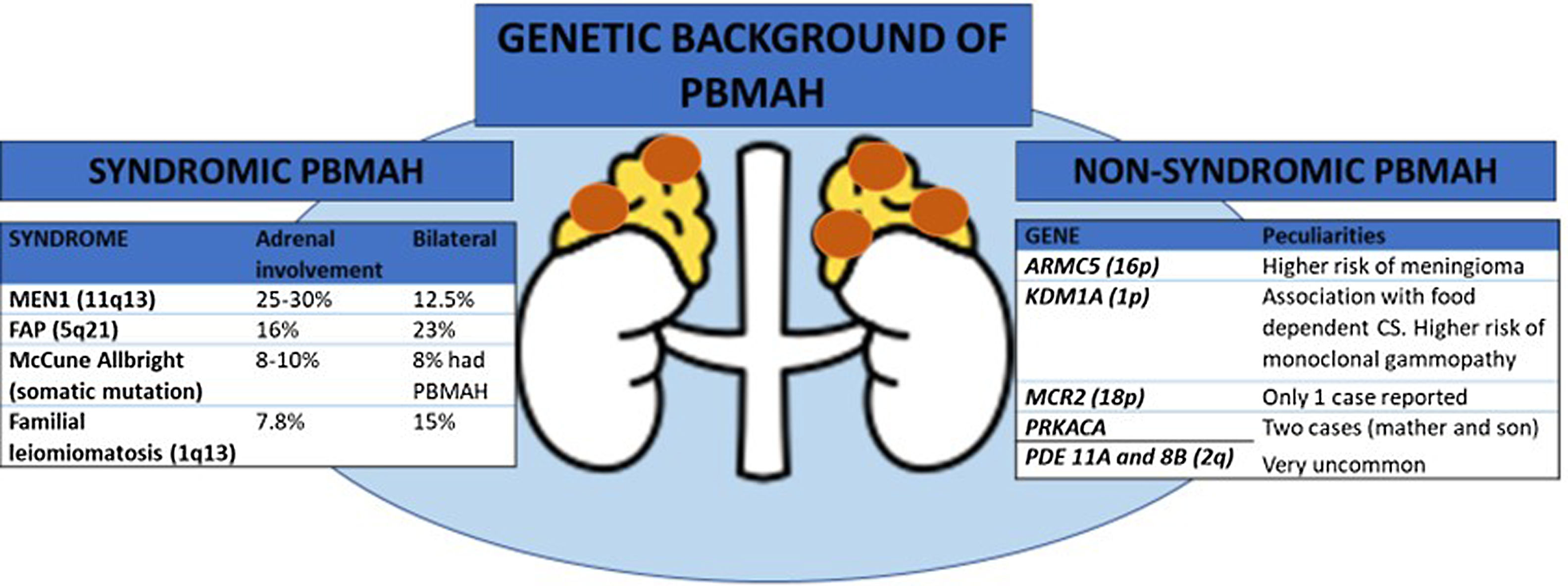
Primary bilateral macronodular adrenal hyperplasia (PBMAH) accounts for less than 1% of all cases of endogenous Cushing's syndrome 1. It is morphologically characterized by bilaterally enlarged adrenal glands containing two or more adrenal nodules larger than 10mm. Although cases of hyperandrogenism and increased aldosterone secretion have been described, increased cortisol production is the most frequent biochemical phenotype. Generally, the disease progresses slowly over time, and often manifests as autonomous cortisol secretion (ACS) with mild hypercortisolism, whereas its presentation as overt Cushing's syndrome is infrequent 1. The bilateral appearance of nodules has put forward the hypothesis of potential germline genetic predisposition to the disease. In fact, the new World Health Organization classification (WHO 2022) of adrenal cortical proliferations highlights the importance of the frequent genetic origin of PBMAH and suggests that the term hyperplasia is not appropriate due to the characteristic clonal and/or neoplastic pattern of the disease. The new term bilateral macronodular adrenal cortical disease is suggested as more appropriate instead of PBMAH 2, but currently lacks endorsement by other scientific organizations.
In many familial cases of PBMAH, the variants in the armadillo repeat containing 5 (ARMC5) gene are closely associated with the disease. The inactivating variants in ARMC5, located on chromosome 16p11.2, are the most common underlying genetic cause of PBMAH 3. Germline pathogenic variants are found in 15% of patients with apparently sporadic PBMAH, 50% of operated patients, and 80% of patients with evident familial presentation 4–6. Several differential features have been described in patients with ARMC5 pathogenic variants compared to those patients harboring wild-type ARMC5, including: i) a more advanced Cushing's syndrome; ii) lower mean age at diagnosis (45-50 vs. 50-55 years); iii) higher adrenal weight and number of adrenal nodules, and iv) a higher prevalence of hypertension 4. Early detection of ARMC5 variant status and familial screening might have important clinical implications leading to an earlier diagnosis of PBMAH. The latter would allow to detect the disease when hypercortisolism is still mild and has not yet generated negative effects on the cardiometabolic system. In addition, pathogenic ARMC5 variants have been associated with a higher risk of intracranial meningiomas 7 (figure 1). Thus, if ARMC5 pathogenic variants are detected, a cerebral MRI should be performed at least at the diagnosis of PBMAH. However, the prevalence of meningioma also seems to be higher in benign adrenocortical adenomas than in age- and sex-matched controls (18% vs. 6%) according to a recent study 8. Nevertheless, the role of ARMC5 in the pathogenesis of sporadic unilateral adrenal adenomas has been poorly investigated, with only non-pathogenetic allelic variants or variants of uncertain significance detected 5.
One of the most important recent discoveries in the field of PBMAH is the role of lysine (K)-specific demethylase 1 A (KDM1A) pathogenic variants in patients with food-dependent Cushing's syndrome 9. KDM1A inactivation appears to be an exclusive genetic mechanism of this phenotype explaining about 90% of cases 9, or even 100%, according to another series 10. Thus, overall, KMD1A pathogenic variants could be present in 10 to 30% of the cases with PBMAH 9. In patients with these pathogenic variants, cortisol secretion is stimulated by food ingestion and diagnosis can be done by analysis of the circadian rhythm of cortisol levels. Moreover, patients with the pathogenic variant are mostly female 9. One important point to consider is that, according to one study, carriers of KDM1A pathogenic variants had a higher risk of developing monoclonal gammopathies. Thus, serum protein electrophoresis should be performed these patients 10 (figure 1).
 ARMC5: armadillo repeat containing 5; CS: Cushing")
Genetic background of primary bilateral macronodular adrenal hyperplasia (PBMAH)
ARMC5: armadillo repeat containing 5; CS: Cushing's syndrome; FAP: familial adenomatosous poliposis; MEN1: multiple endocrine neoplasia type 1, MCR2: adrenocorticotropic hormone receptor; PRKACA: protein regulatory subunit Kinase A; PDE11A: phosphodiesterase 11A; PDE8B: phosphodiesterase 8B; KDM1A: lysine (K)-specific demethylase 1 A; PBMAH: primary bilateral macronodular adrenal hyperplasia.
All the genes associated with PBMAH that have been identified are transmitted in an autosomal dominant mode. Moreover, for constitutive ARMC5 variants, the penetrance of the disease is quite high, probably above 80–85% in adults above 45 years, but with variable severity of cortisol dysregulation between relatives 11. Hence, genetic screening for ARMC5 and KDM1A pathogenic variants can now be offered to most PBMAH patients and their families, opening the way to an earlier diagnosis of hypercortisolism and potentially associated comorbidities and extraadrenal malignancies. This could lead to an optimization in their management. Nevertheless, pathogenic variants in ARMC5 have not been reported in patients with food dependent Cushing's syndrome, making ARMC5 and KDM1A pathogenic variants mutually exclusive. Therefore, investigating ARMC5 might not be necessary in these patients 9. Moreover, a recent study suggests that genotyping of ARMC5 should be limited to patients with clear bilateral adrenal involvement and ACS since this combination holding a 100% sensitivity for detecting ARMC5 pathogenic variants 6. Beyond ARMC5 and KDM1A pathogenic variants, patients with PBMAH may occasionally - approximately 5% of the cases- present alterations in genes responsible for a multiple tumor syndromes, including multiple endocrine neoplasia type 1 (MEN1), adenomatous polyposis coli (APC), and fumarate hydratase (FH) genes 1. In addition, pathogenic variants in the adrenocorticotropic hormone receptor (MCR2)12, protein regulatory subunit Kinase A (PRKACA gene) 13, phosphodiesterase 11A (PDE11A gene) 14 and 8B (PDE8B gene) 15, have been anecdotally described as a cause of PBMAH (see figure).
In conclusion, the identification of frequent pathogenic variants in ARMC5 and KDM1A genes in patients with PBMAH, including familial and apparently sporadic cases, indicates that it is a disorder with a very common genetic origin. Thus, genetic studies should be considered in all patients with PBMAH with the aim to achieve an earlier diagnosis of hypercortisolism and optimize its management. Testing for ARMC5 is not indicated in patients with food dependent Cushing's syndrome, while KDM1A should be the first gene to study in these patients.
Ethical approvalNot applicable
Financial SupportNot applicable
Conflict of interestThe authors have no conflict of interest
