


INTRODUCCIÓN
Los tumores de la corteza suprarrenal suponen una enorme rareza en la infancia, como se describe en la bibliografía1 (un 0,4% de los tumores malignos de la infancia). En general, son activos desde el punto de vista hormonal, y producen cuadros de virilización posnatal en los casos en que la testosterona está elevada o, más raramente, clínica de feminización. En general, los tumores suprarrenales funcionantes son más frecuentes en las mujeres, sin que actualmente se sepa la razón de este hecho. En los casos con virilización, en el varón darán lugar a una seudopubertad precoz isosexual y en la mujer, masculinización. En el caso de tumores suprarrenales feminizantes, muy raros aunque más frecuentes en los varones, el motivo de consulta suele ser ginecomastia en los chicos y seudopubertad precoz isosexual en la mujer.
Los tumores suprarrenales no funcionantes son muy raros antes de los 20 años de edad. En estos casos el motivo de consulta más frecuente sería el de una masa abdominal, anorexia o más raramente fiebre.
Hasta la fecha, el tratamiento de elección es quirúrgico. La quimioterapia con Op'-DDD sólo se administra en casos muy seleccionados de mal pronóstico.
En este trabajo se describe un caso de carcinoma suprarrenal hormonalmente activo por un aumento de producción de testosterona. El interés de esta publicación se centra en la rareza de este cáncer, y en la predisposición genética para presentarlo junto con otros tumores entre miembros de una misma familia, así como su asociación frecuente a otras enfermedades no neoplásicas (hemihipertrofia, síndrome de Beckwith-Wiedemann), sin que exista un nexo filontogenético que las relacione. Finalmente, es importante describir las modalidades terapéuticas existentes, resaltando las dificultades presentes por carecer de un tratamiento más eficaz en el 100% de los pacientes.
CASO CLÍNICO
Se presenta a una niña, actualmente con 17 años, que a los 10 meses de vida es remitida al hospital por presentar vello pubiano y aumento del tamaño del clítoris, observado a partir de los 6 meses de vida y que había aumentado notablemente en las últimas semanas. No existían otros signos puberales, y la velocidad de crecimiento era normal. No había tratamiento hormonal pre o posnatal.
Era hija de padres sanos, no consanguíneos, con tallas familiares medias y sin antecedentes familiares de pubertad precoz, muertes neonatales o genitales ambiguos. Tenía un hermano mayor sano. El segundo hermano había sido intervenido a los 2 años de vida de rabdomiosarcoma embrionario testicular izquierdo, y en la actualidad está asintomático. El tercer hermano falleció a los 8 años de un tumor de plexos coroideos.
Exploración física (primera consulta)
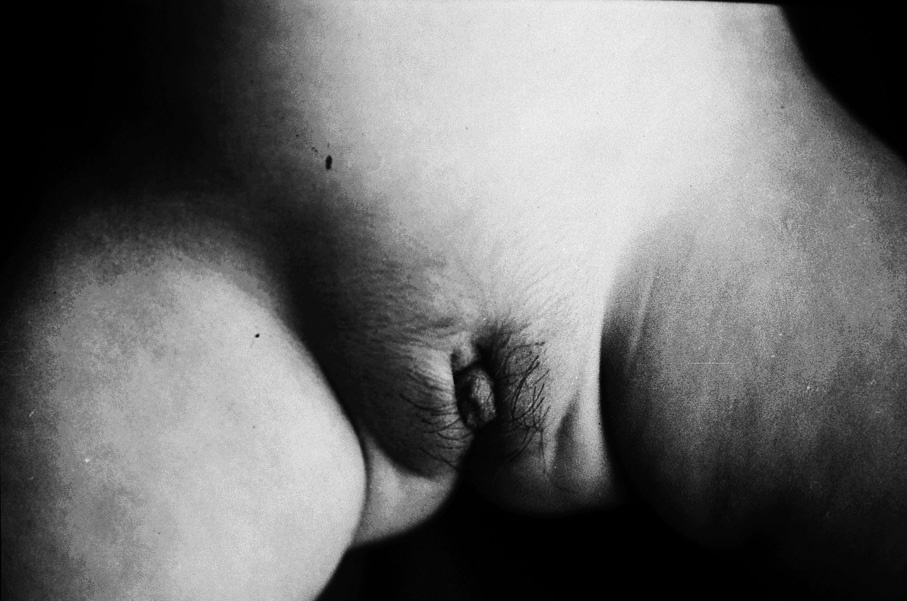
El peso y la talla se situaban en +1 desviación estándar (DE). No había signos de hipercortisolismo. La presión arterial era normal. No se observaban malformaciones aparentes. En la zona abdominal, se palpaba una masa en la fosa ilíaca izquierda (FII), bien delimitada, de tamaño inicial de 4 * 4 cm (que llegó a alcanzar en los días previos a la intervención 8 * 6 cm). No había visceromegalias. Los genitales externos estaban normopigmentados y presentaba un falo de 2 * 1,5 cm, aproximadamente. Había sinequia de labios menores y vello en los labios mayores, largo y rizoso. No se palpan gónadas. No había axilarquia, telarquia ni cambios de sudoración (fig. 1).
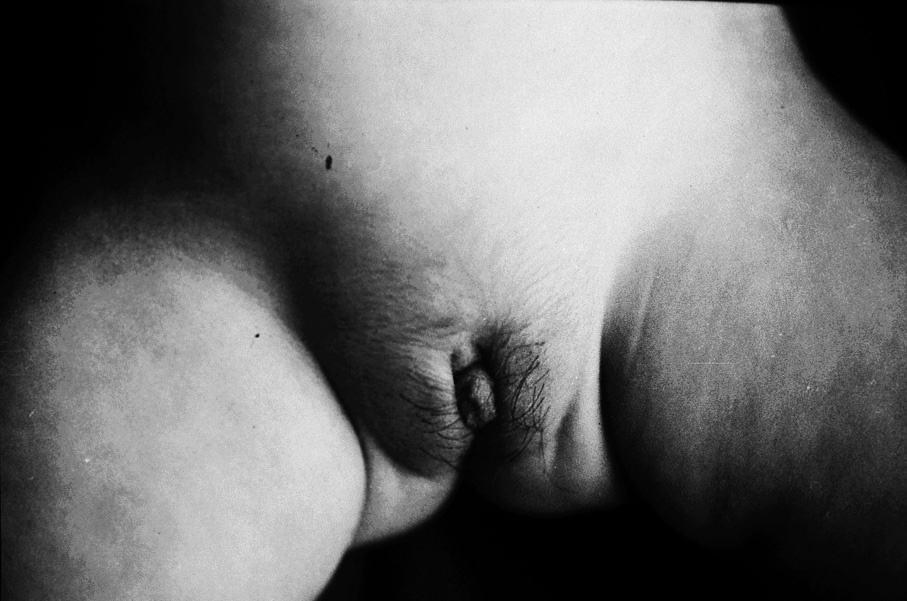
Fig 1. Lactante de 10 meses, mujer, con estigmas de seudopubertad precoz isosexual (genitales ambiguos, virilización): clítoris hipertrófico (falo); pubarquia (labios mayores); sinequia de labios menores, y pigmentación normal.
Exploraciones complementarias realizadas
Analítica basal: hemograma: anemia (hemoglobina: 10,2 g/dl). Sustancia blanca y plaquetas: normal. Velocidad de sedimentación globular (VSG), 45. Bioquímica: GOT, 59; GPT, 67. Resto normal.
Cariotipo: 46XX normal.
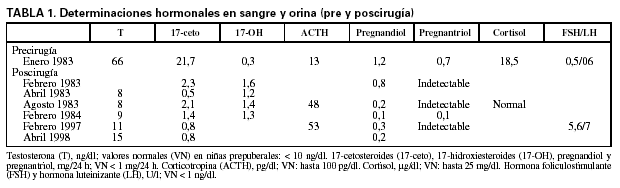
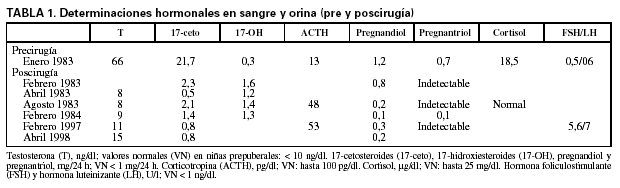
Determinaciones hormonales en sangre y orina (pre y poscirugía) (tabla 1).
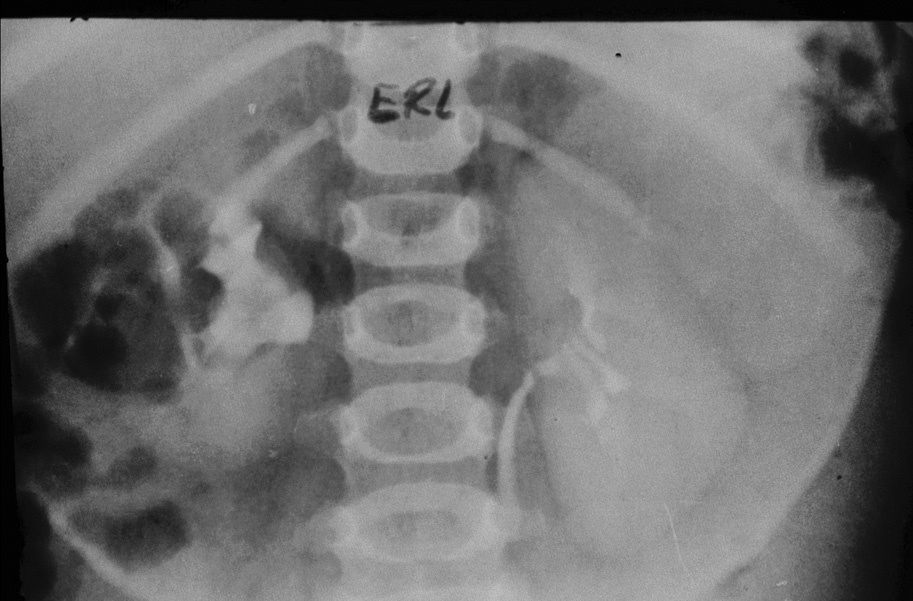
Estudio radiológico: a) ecografía abdominal: masa de características ecográficas sólidas con zonas hiperecogénicas que pueden corresponder a calcio, de tamaño 8 * 6 *4 cm, independiente de riñón y bazo, y que corresponde a la zona suprarrenal, y b) urografía por vía intravenosa (fig. 2).
Fig. 2. A los 15 min, se delimita una masa hipoecogénica, situada en la zona anterior del riñón y el bazo.
Evolución
Fue intervenida a los 10 días de su ingreso, con cobertura hormonal perioperatoria, y se extirpó una masa grande, bien delimitada en la zona suprarrenal, con el diagnóstico de presunción de carcinoma suprarrenal virilizante, que posteriormente se confirmó por el estudio anatomopatológico.
Dado que este tipo de tumores son radiorresistentes, no se administró radioterapia. Ante la ausencia de metástasis y dada la alta toxicidad del fármaco más indicado en estos casos (Op'-DDD), tampoco se trató con quimioterapia.
Desde la intervención quirúrgica hasta la actualidad, la paciente ha sido controlada periódicamente, y no se ha objetivado ninguna alteración de la secreción hormonal. Ha mantenido un ritmo de crecimiento en el límite alto de la normalidad (talla, +2 DE), con una edad ósea acorde con su edad cronológica. La talla final (edad cronológica, 20 años) es de 170,5 cm (+1 DE, +2 DE). Pronóstico de talla genética (PTG): 157 ± 5 cm (media, 2 DE). El psiquismo es normal para su edad. Todas las exploraciones realizadas (ecografía y tomografía computarizada [TC] abdominal) han sido normales.
MÉTODOS Y COMENTARIOS
El interés de esta publicación se centra en 4 aspectos fundamentales:
1. Es importante resaltar la rareza de este tipo de tumores en la infancia (2 por cada 105 cánceres pediátricos). Como se describe en la bibliografía, la mayor parte de los carcinomas suprarrenales que se presentan en la edad pediátrica son funcionalmente activos, factor que favorece su diagnóstico temprano. Considerando esta capacidad secretora de hormonas (que no guarda relación con el tipo histológico) se pueden diferenciar varias formas de presentación. La virilización, en tumores secretores de andrógenos, es la forma clínica más frecuente (66%), seguida por el hipercortisolismo (30%), en el seno de tumores secretores de glucocorticoides. Habitualmente, este tipo de tumores suelen ser mixtos, y también segregan andrógenos y/o mineralocorticoides. La feminización, como motivo de consulta, o el hiperaldosterismo, como sintomatología más compleja, son excepcionales. En adultos, sin embargo, este tipo de tumores suele comenzar por efecto masa, y en general son hormonalmente inactivos. Por ello, suelen diagnosticarse de forma tardía, cuando se asocian metástasis al carcinoma primario y, en consecuencia, el pronóstico empeora1-2.
2. Otro aspecto interesante de este tipo de tumores es la relación que existe entre el carcinoma suprarrenal y algunas enfermedades no neoplásicas. Como ocurre en el caso de la hemihipertrofia congénita3, el síndrome de Beckwith-Wiedemann4,5; defectos genitourinarios, hamartomas y alteraciones del sistema nervioso central (SNC). De hecho, en todas estas enfermedades se describe un aumento de la incidencia de este tipo de tumores respecto a la población sana de control.
3. Cabe subrayar, además, la predisposición genética para presentar carcinoma suprarrenal y otras neoplasias, tanto en los propios enfermos como en sus familiares. Hasta tal punto, que se puede hablar de "familias con predisposición para el cáncer"6(cancer prone families), término descrito por Li y Fraumeni para describir a las familias en las que se observa en diversos hermanos la asociación de tumores de estirpe neural (rabdomiosarcomas y carcinoma suprarrenal). Últimamente, también se ha descrito una incidencia aumentada de asociación con el tumor de Wilms y los hamartomas. Es importante añadir, que estos tumores pueden aparecer en un mismo miembro de la familia como tumor secundario o primario.
En nuestro caso, pudimos comprobar, en el momento del diagnóstico de carcinoma suprarrenal, cómo se cumplía esta predisposición familiar descrita en la bibliografía, puesto que el segundo hermano ya había sido diagnosticado de rabdomiosarcoma testicular. Además, durante los 9,5 años posteriores de seguimiento, pudimos asistir a la aparición de un nuevo tumor primario en el seno de esta familia: el tercer hermano, a los 7 años presentó un cuadro de hipertensión intracraneal, y fue diagnosticado de carcinoma de plexos coroideos de evolución fatal.
De esta asociación frecuente, descrita ya en varios casos en la bibliografía, se obtienen 2 conclusiones:
La identificación temprana de cada uno de los miembros afectados de estas familias "con predisposición para el cáncer" tiene un enorme interés desde el punto de vista tanto terapéutico como preventivo.
Esta asociación descrita sugiere la existencia de factores oncogénicos que probablemente intervendrían durante el desarrollo embrionario8. De hecho, en la bibliografía ya hay varios estudios que describen fundamentalmente 2 anomalías genéticas que podrían favorecer el desarrollo de este tipo de tumores. Una sería la alteración del gen del factor de crecimiento similar a la insulina 2 (IGF-2), presente en la región cromosómica del 11p 13-15'. Gicquel et al describieron la sobreexpresión del gen de la IGF-2 predominante en pacientes con carcinoma suprarrenal frente a pacientes con adenoma suprarrenal, lo que según estos autores se podría considerar un indicativo de malignidad9 (alteración genética presente en 12 tumores suprarrenales malignos y en 2 adenomas suprarrenales de un total de 23 tumores suprarrenales benignos).
La segunda anomalía genética descrita en estos pacientes es la falta de inhibición del oncogén p53 (gen supresor de tumores) situado en 17p13. Esta anomalía, de presentarse en familias con algún caso de carcinoma suprarrenal, condicionaría la aparición de otros tumores (rabdomiosarcomas, osteosarcomas, cáncer de mama)10. Además, algunos autores postulan esta anomalía cromosómica como un claro indicador de malignidad en caso de presentarse en un paciente con tumor suprarrenal11, como Gic et al que, en una serie de 28 tumores suprarrenales, observan anomalía positiva en todos los carcinomas11 frente a sólo 2 de 17 adenomas.
4. Por último, otro aspecto polémico en relación con estos tumores son las últimas tendencias terapéuticas. Para todos los autores, el tratamiento de elección es la extirpación quirúrgica completa y temprana, intentando mantener la cápsula intacta. Esta cirugía se debe realizar siempre con tratamiento corticoideo sustitutivo (aunque el tumor no sintetice glucocorticoides), por la posible situación de hipocortisolismo transitoria que, en principio, puede sobrevenir con la cirugía. Si el tumor es bilateral y se van a extirpar ambas glándulas, el tratamiento corticoideo sustitutivo se mantendrá de por vida.
El principal problema en este tipo de tumores malignos es que la mayoría de las veces, cuando se realiza el diagnóstico, el paciente se encuentra en un estadio avanzado (metástasis locales o a distancia). Por ello, se impone la necesidad de asociar otros tratamientos a la cirugía.
La radioterapia no ha demostrado su eficacia como tratamiento paliativo y, asociada a la exéresis microscópica incompleta, tampoco evita la recidiva local.
En la actualidad, la primera línea quimioterápica recomendada en pacientes con tumores irresecables o en los que la cirugía está contraindicada, sigue siendo el mitotane (Op'-DDD). Ya hay varios estudios que confirman una respuesta clínica positiva del tumor tras el uso de este fármaco. Entendiendo "respuesta clínica positiva" como la disminución del tamaño o la desaparición completa del tumor por métodos demostrables (TC, confirmación por recirugía o autopsia). La mayoría de estos estudios coinciden en que la tasa de respuesta tumoral con este fármaco es de alrededor del 30% (de pacientes no curados con cirugía a los que se les ha administrado mitotane) y que es preciso alcanzar unos valores séricos adecuados para que el fármaco sea efectivo. Así, Van Slooten et al12 no obtienen efectos terapéuticos significativos con valores séricos de Op'-DDD menores de 10 µg/ml. Mientras que con valores en suero mayores de 14 µg/ml, observaron un incremento de la supervivencia y/o una regresión tumoral hasta en un 57% de los pacientes (en 8 pacientes de un total de 14 carcinomas suprarrenales). Por desgracia, el empleo de este fármaco es bastante complicado, dada la gran cantidad de efectos adversos y la toxicidad que provoca su administración, especialmente en niños. Los efectos secundarios más frecuentes suelen ser vómitos, anorexia y los producidos por la toxicidad sobre el SNC (ataxia, somnolencia, anomalías del electroencefalograma e incluso daño cerebral grave)13. Así, la mayoría de autores coinciden en que la dosis máxima tolerada en niños es de 6 g/m2 s.c./día.
Finalmente, es necesario añadir que la duración media de la respuesta terapéutica tras administrar mitotane oscila entre los 7 y los 12 meses en adultos. Sin embargo, a pesar de que responde un 30% de pacientes, muy pocos estudios describen una respuesta prolongada con el Op'-DDD (remisión completa o duradera en el tiempo). Se han ensayado asociaciones con el mitotane para mejorar su eficacia, aunque los resultados han sido bastante descorazonadores. Así, las asociaciones del mitotane más utilizadas han sido con 5-fluorouracilo, cisplatino o estreptozocina14-16.
En resumen, y teniendo en cuenta la revisión realizada en este trabajo, concluimos que ante un carcinoma suprarrenal la actitud terapéutica inicial es siempre la cirugía. En caso de que la extirpación no pueda llevarse a cabo de forma completa o el paciente se encuentre en un estadio avanzado de la enfermedad, la opción de asociar quimioterapia, en concreto Op'-DDD, debe valorarse de forma individualizada y, en conjunto, por los distintos especialistas que traten al niño (oncólogos, endocrinólogos, cirujanos).
