


INTRODUCCIÓN
El síndrome de McCune-Albright (SMA) se describió en la década de los treinta del siglo pasado y se define clásicamente como la asociación de lesiones hiperpigmentadas cutáneas únicas o múltiples de color café con leche, planas y con bordes irregulares, pubertad precoz y displasia fibrosa1-6.
Los pacientes con SMA pueden presentar, también, otras anomalías endocrinas y no endocrinas. Algunos pacientes presentan sólo 2 de los hallazgos de la tríada clásica o 1 y otra anomalía, endocrina o no endocrina4,5.
Dentro de las alteraciones de las glándulas endocrinas podemos encontrar tirotoxicosis, bocio, síndrome de Cushing, acromegalia, hiperprolactinemia, hiperparatiroidismo y raquitismo hipofosfatémico hiperfosfatúrico5-7. La endocrinopatía más frecuente es la pubertad precoz, que es isosexual; la segunda más frecuente es el hipertiroidismo2,6.
Por lo que respecta a la afección ósea, cualquier hueso puede verse afectado, aunque las localizaciones más frecuentes son los huesos craneofaciales, las costillas, el fémur y la tibia2.
El SMA ocurre esporádicamente y no aparece en las generaciones siguientes. Se debe a una mutación que activa la subunidad * de la proteína G que conduce a una hiperactividad de la adenilciclasa en diversos órganos1-4,8,9.
Presentamos el caso de una mujer con SMA como paradigma de afección tumoral en múltiples glándulas endocrinas.
CASO CLÍNICO
Mujer de 42 años diagnosticada de displasia fibrosa poliostótica desde su juventud, con múltiples fracturas patológicas desde la infancia, que han ocasionado deformidades y requerido múltiples intervenciones quirúrgicas.
La paciente nació de parto normal, con un peso aproximado de 4.000 g, y recibió lactancia materna. Ya desde el nacimiento llamaba la atención la presencia de múltiples manchas café con leche, de bordes dentados en la parte posterior del cuello y la espalda. Desde los 13 meses de edad presentó hemorragias vaginales de duración y cuantía variable, con menstruaciones regulares desde los 7 años de edad. También refiere aceleración del crecimiento, con una talla final de 142 cm.
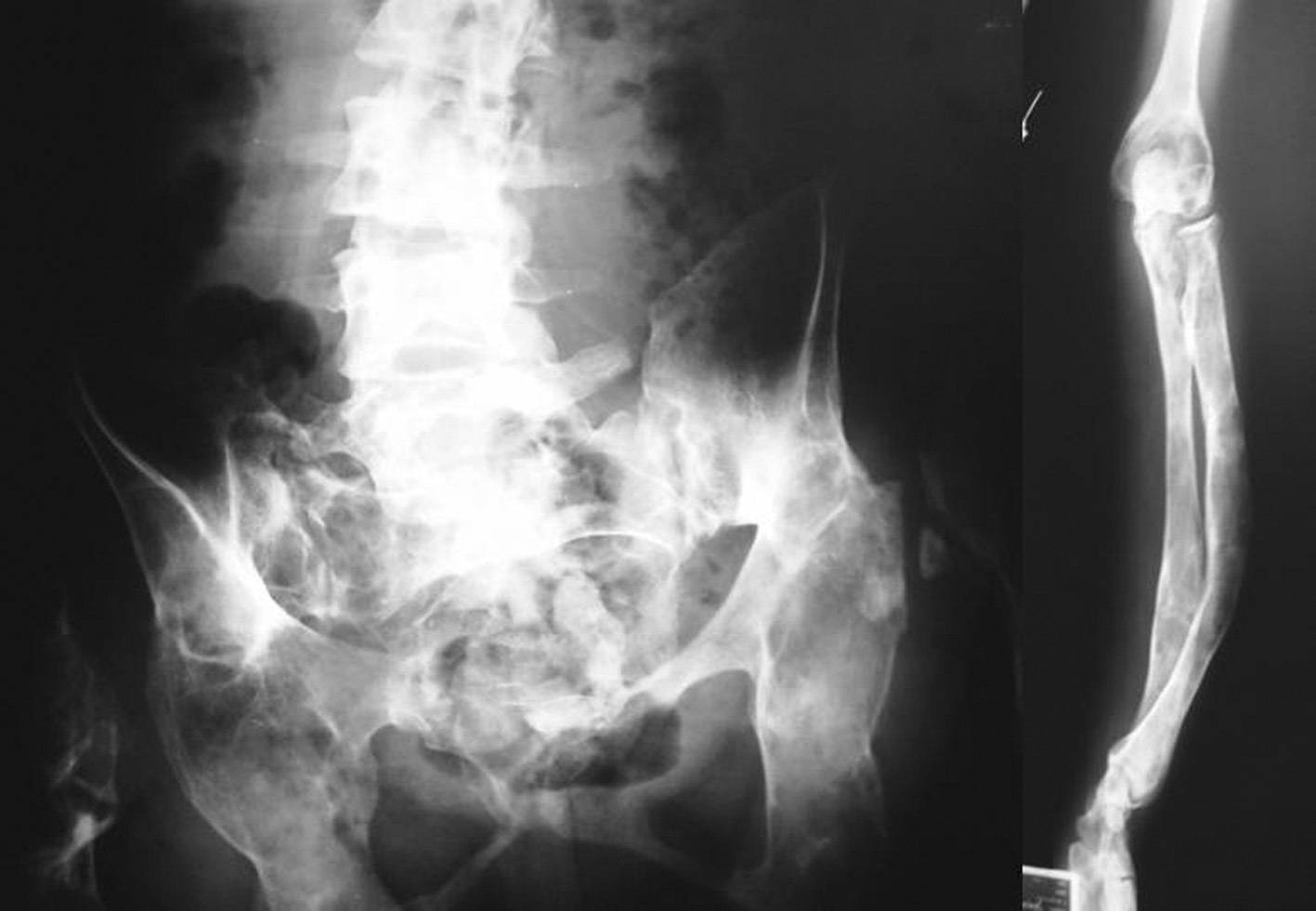
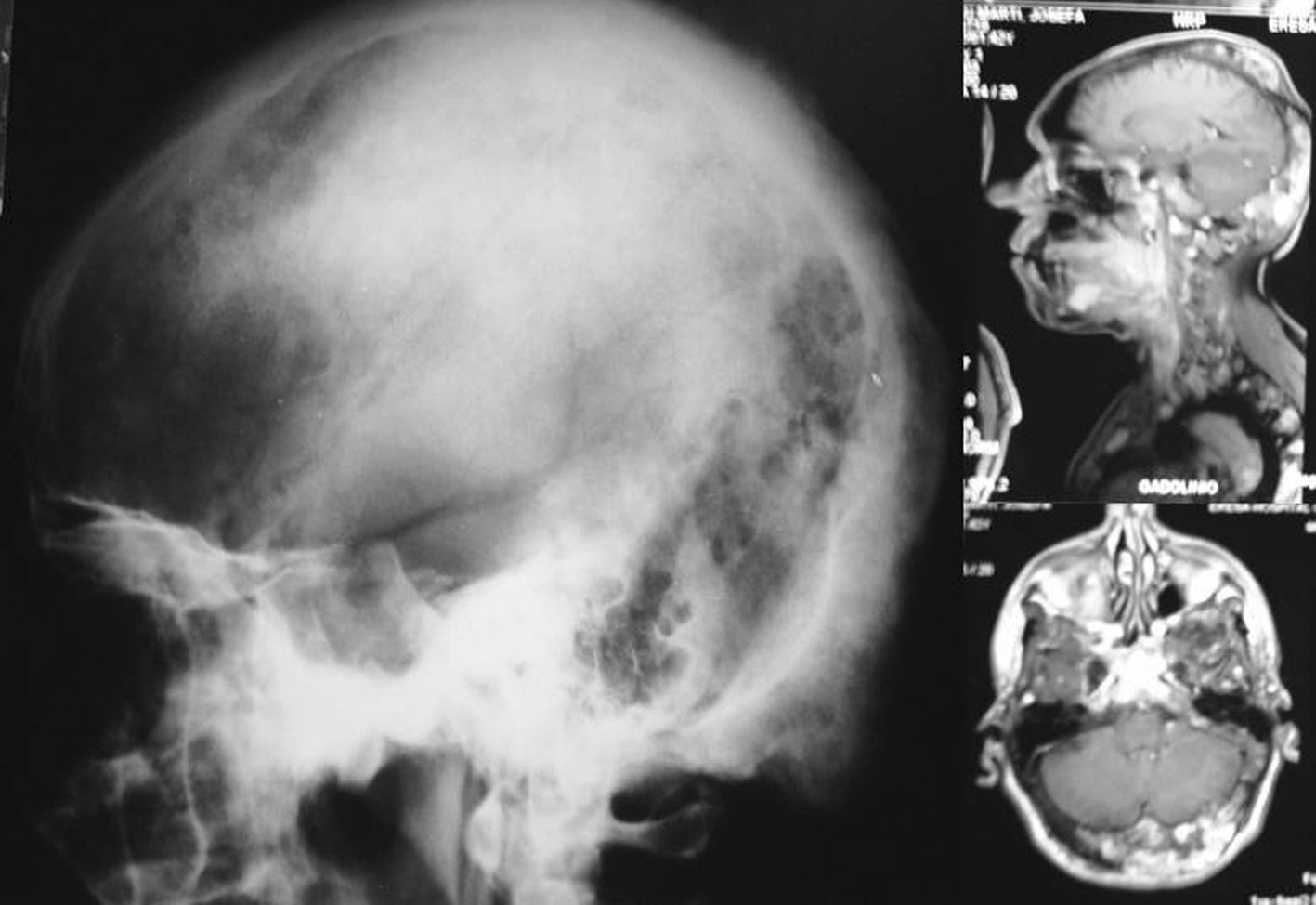
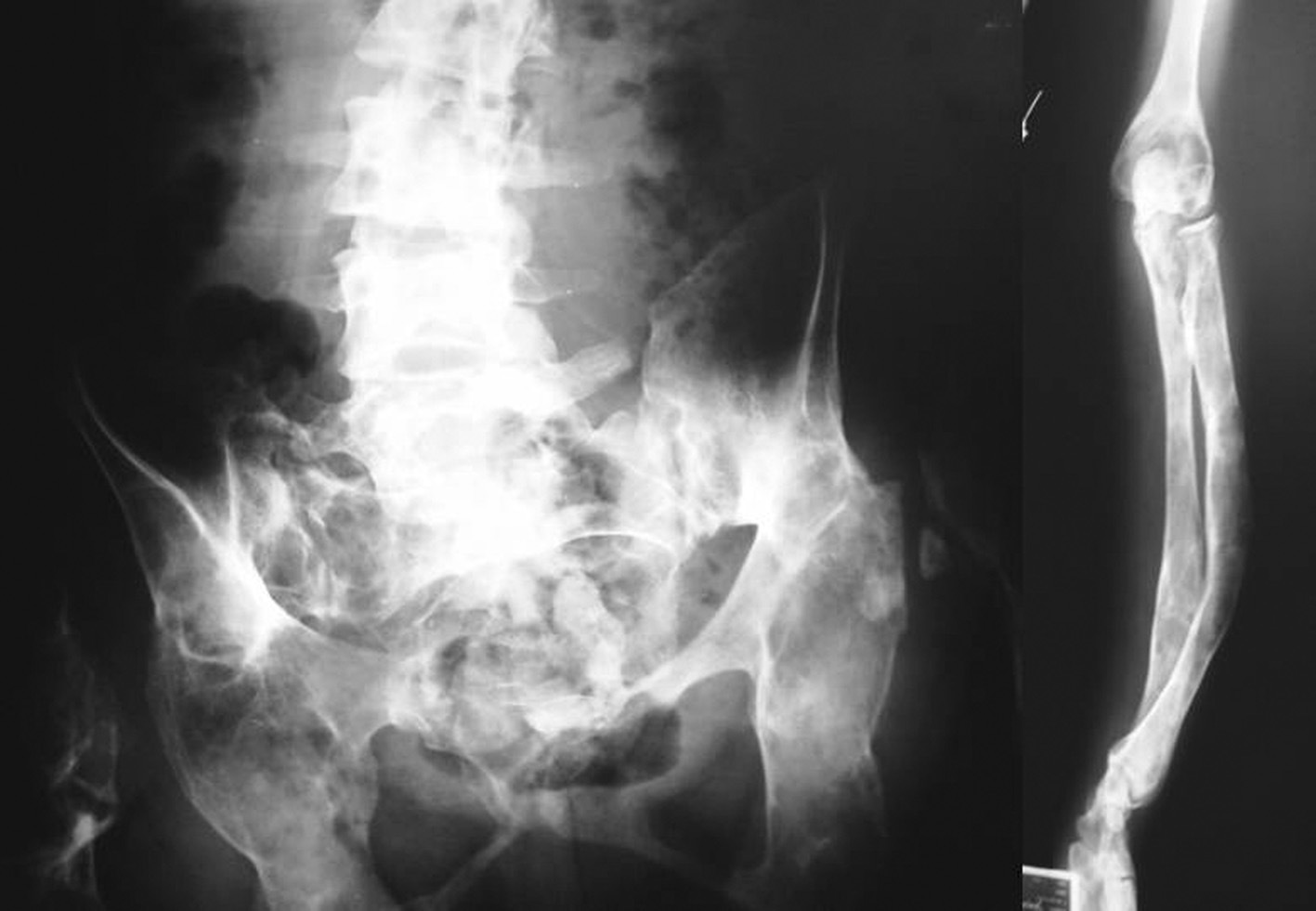
Desde la infancia presentó múltiples fracturas patológicas que ocasionaron deformidad en ambas caderas, las tibias y el antebrazo izquierdo, lo que obligó a practicar varias intervenciones quirúrgicas: osteosíntesis de la cadera derecha a los 10 años de edad, osteosíntesis de la cadera izquierda a los 11 años, osteotomía de la pierna izquierda en 2 ocasiones, a los 12 y a los 24 años. Existía también hiperostosis cortical en el cráneo. En las figuras 1 y 2 se presentan algunos de los hallazgos radiológicos óseos. Además, presentó 2 episodios de pancreatitis aguda durante su juventud.
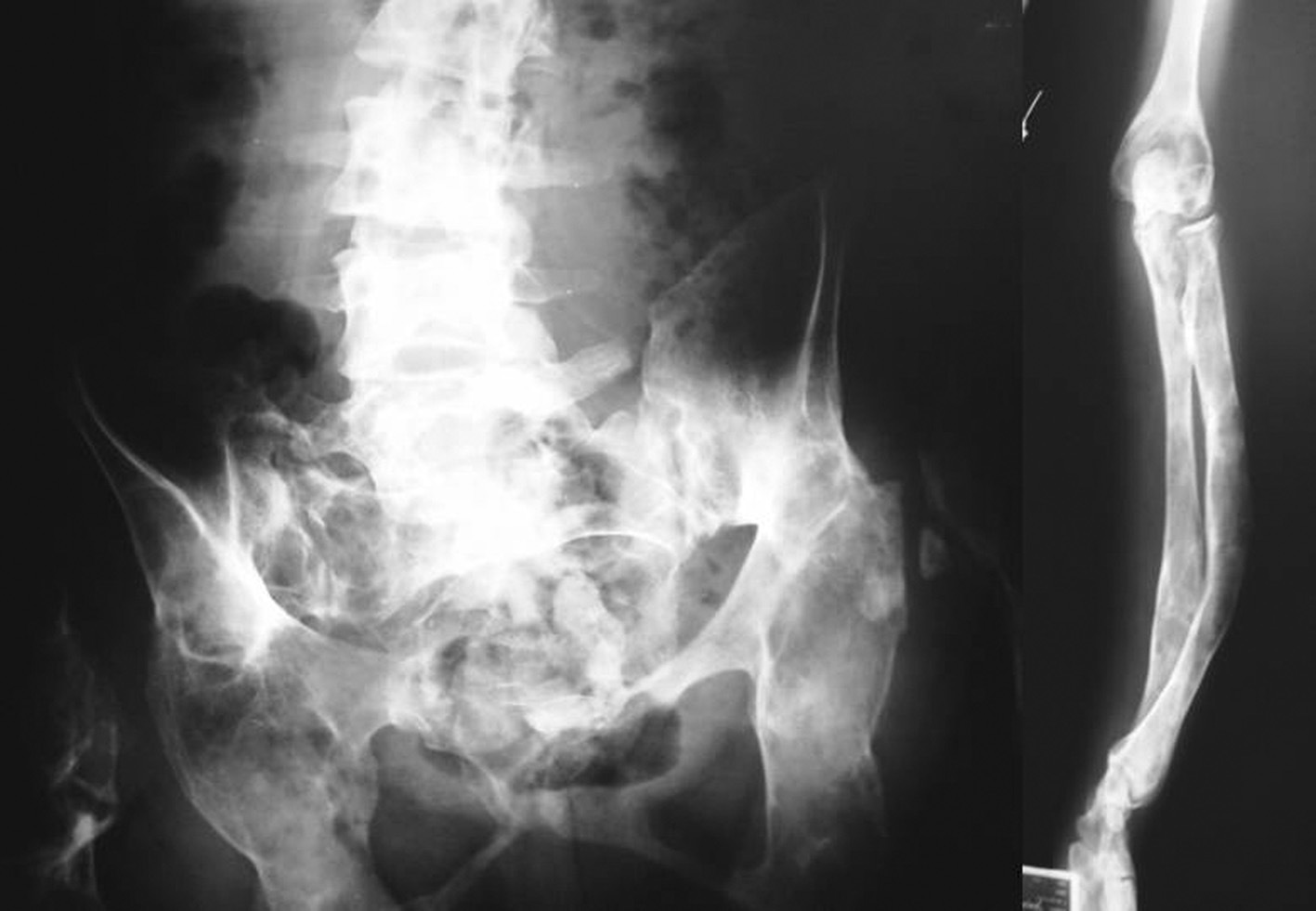
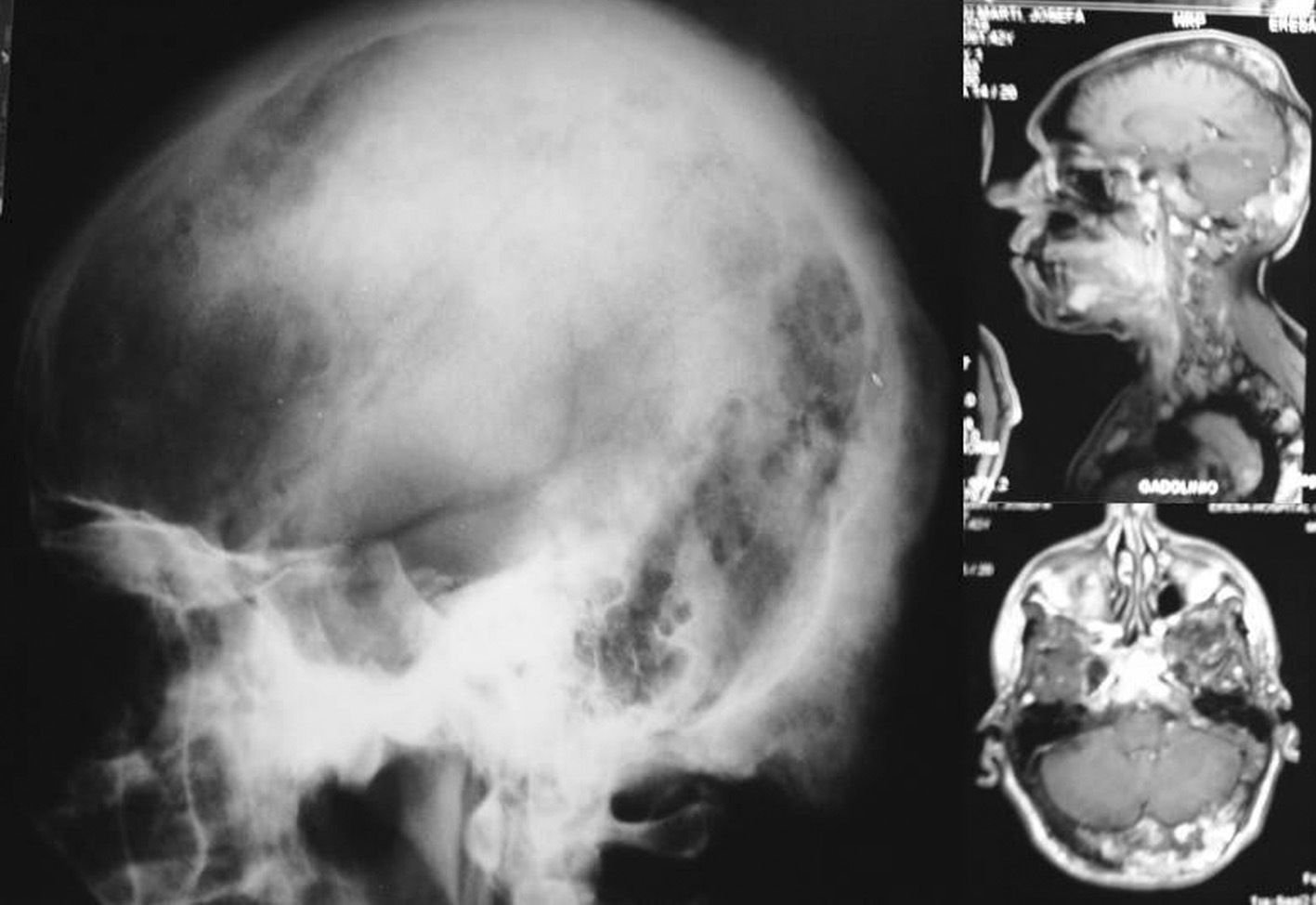
Fig. 1. En la radiología convencional (izquierda) y en la resonancia magnética (derecha) se observan las deformidades en el cráneo.
Fue vista por primera vez en el Servicio de Endocrinología de nuestro hospital a los 25 años de edad, remitida por el Servicio de Traumatología, al aparecer durante la última intervención quirúrgica presentó un episodio de taquicardia sinusal (180 lat/min) y clínica compatible con hipertiroidismo. Interrogada, la paciente refirió que desde hacía aproximadamente 4 meses sentía un temblor fino de manos, hipersudoración, intolerancia al calor, taquicardia y palpitaciones, sin pérdida de peso, insomnio ni alteraciones del ritmo defecatorio.
En la exploración física presentaba una talla de 142 cm y un peso de 59 kg, piel grasa, sudorosa y caliente. Se observaba un aumento del tamaño de la glándula tiroides a expensas del lóbulo izquierdo, de superficie irregular. En la auscultación cardíaca, los tonos eran rítmicos a 110 lat/min, sin soplos, y la presión arterial era de 140/90 mmHg.
En el análisis practicado destacó una tiroxina (T4) libre de 13,6 ng/dl y una triyodotironina (T3) total de 3,42 ng/dl, con tirotropina (TSH) suprimida (0,01 µU/ml). Los anticuerpos antitiroideos fueron negativos. Se realizó ecografía cervical que objetivó un lóbulo izquierdo de gran tamaño, con áreas anecoicas. La gammagrafía tiroidea mostró una glándula aumentada de tamaño a expensas del lóbulo izquierdo, con captación heterogénea, que alternaba zonas de captación intensa y disminuida, todo ello compatible con bocio multinodular. Se procedió a tratamiento con 17,5 mCi de 131I, seguido de una disminución del tamaño del bocio; la paciente permaneció clínica y analíticamente eutiroidea en todos los controles posteriores.
Ocho años después de ser vista por primera vez en nuestro servicio, la paciente dejó de acudir a los controles. Durante ese tiempo presentó galactorrea y, tras una biopsia mamaria, fue diagnosticada de carcinoma papilar in situ, por lo que se procedió a una mastectomía izquierda a los 36 años de edad en otro centro.
Tras 6 años sin seguimiento por nuestra parte, acudió al servicio de urgencias, donde fue diagnosticada de diabetes mellitus tipo 2, y reinició los controles en la consulta externa de Endocrinología y Nutrición de nuestro centro. Un año después fue diagnosticada de hiperparatiroidismo primario; en el análisis destacaba: calcio, 11 mg/dl; fósforo, 2,9 mg/dl; parathormona (PTH) intacta, 161 pg/ml (valores normales, 10-55); proteínas totales, 7,1 mg/dl; calciuria, 654 mg/24 h, y fosfaturia, 848 mg/24 h. Se realizó una gammagrafía paratiroidea, donde no se observó ninguna lesión que sugiriera adenoma paratiroideo, y una resonancia magnética, que resultó negativa.
Además, se observó que presentaba aspecto acromegaloide, por lo que se realizó una determinación de la hormona de crecimiento (GH) basal, que fue de 9,4 ng/ml, y del factor de crecimiento similar a la insulina tipo 1 (IGF-1), que fue de 661 ng/ml (valores normales, 123-463). La sobrecarga oral con 75 g de glucosa objetivó una GH de 5,6 ng/ml posfrenación, por lo que la paciente fue diagnosticada de acromegalia en actividad. Por ello, se realizó resonancia magnética hipotálamo-hipofisaria, donde se observó una glándula hipofisaria asimétrica con aumento de volumen del lóbulo derecho, con expansión hacia el seno cavernoso; se apreció, además, en el lóbulo izquierdo, una lesión nodular hipocaptante de aproximadamente 3-4 mm. El estudio de las otras hormonas hipofisarias en situación basal y tras pruebas de estimulofrenación fue normal. La paciente estaba pendiente de tratamiento quirúrgico del hiperparatiroidismo primario y de la acromegalia, pero falleció poco después, a los 42 años de edad, por una neumonía adquirida en la comunidad de etiología no filiada.
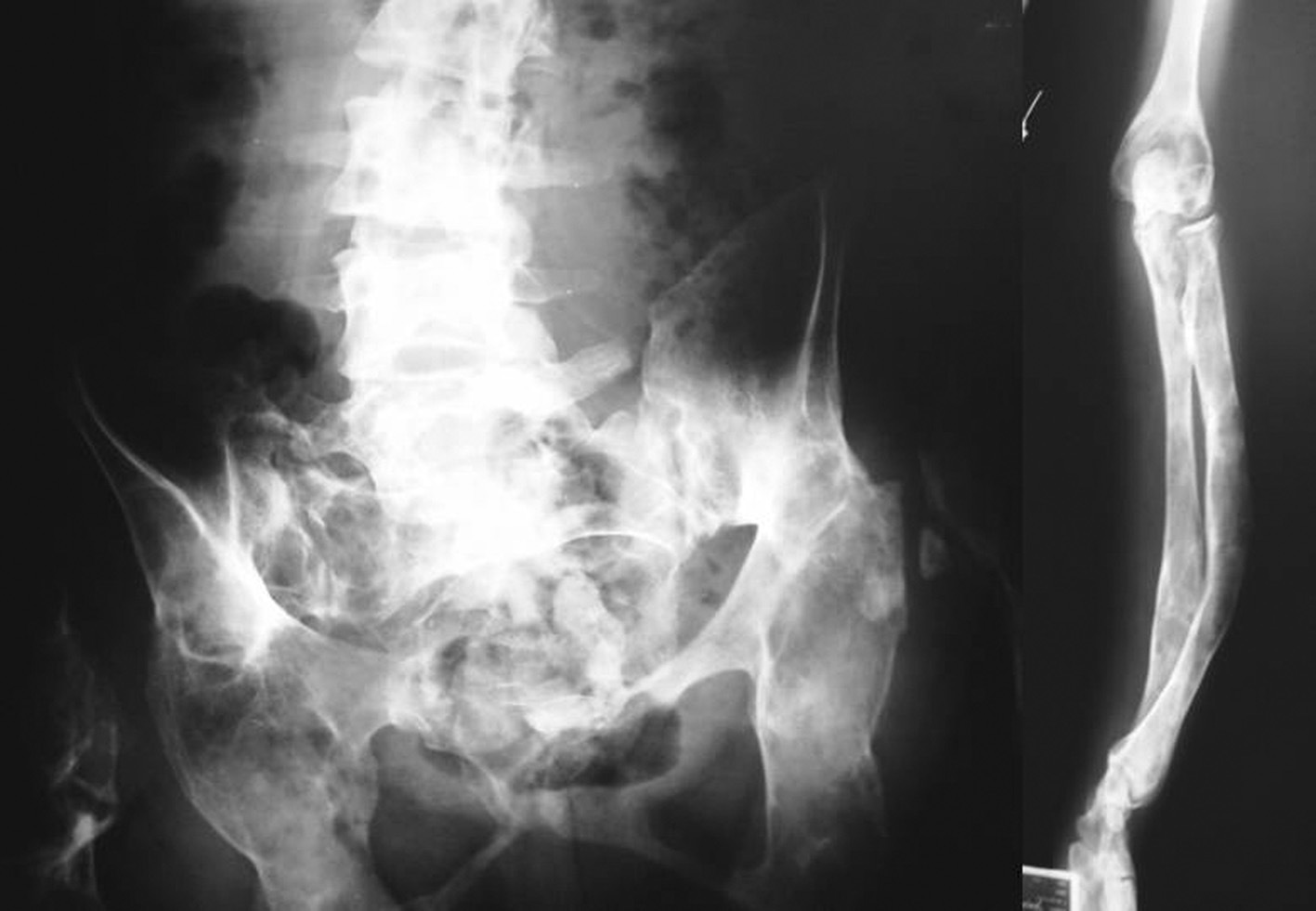
Fig. 2. Deformidades óseas presentadas por la paciente en la pelvis, la columna vertebral lumbosacra y el antebrazo.
DISCUSIÓN
El SMA es una enfermedad infrecuente, más prevalente en las mujeres2. Representa uno de los espectros fenotípicos causados por una mutación activadora en el gen GNAS 1, que codifica la proteína Gs*. Se trata de una mutación poscigótica en la posición 201 de dicha proteína, que genera una deficiencia intrínseca de guanosín trifosfato (GTP) que conduce a una activación persistente de la producción de adenilciclasa y adenosín monofosfato (AMP) cíclico. Ello estimula la proliferación y la secreción hormonales, y puede originar tumores endocrinos diferenciados con incremento en la tasa de liberación hormonal. Cada una de estas glándulas endocrinas periféricas funciona de forma autónoma, al margen de su respectiva hormona hipofisaria1-4,8,9.
El SMA es una enfermedad esporádica, debida a una mutación en las células somáticas durante el desarrollo embrionario. Se trata de mutaciones estructurales que constituyen mosaicos y, por tanto, no están presentes en todas las células. Las que descienden de la célula mutada pueden dar lugar a manifestaciones propias del SMA, mientras que las procedentes de células no mutadas desarrollan tejidos normales. El momento del desarrollo en el que acontece la mutación determina el número de tejidos afectados y la gravedad de la expresión4. Así, las mutaciones tempranas conducen a manifestaciones "floridas" mientras que las tardías suponen una enfermedad más focal. Generalmente, las anomalías en el SMA están restringidas al hueso, la piel y los órganos endocrinos; por tanto, afectan poco a la mortalidad. Sin embargo, algunos pacientes también desarrollan una o más anomalías no endocrinas que pueden aumentar de forma marcada la morbimortalidad1,2,7. En el caso presentado no se pudo estudiar la base molecular del SMA.
Nuestra paciente cumplía los criterios diagnósticos: presentaba la tríada clásica (manchas café con leche, pubertad precoz y displasia fibrosa poliostótica), junto con otras manifestaciones endocrinas: hipertiroidismo primario por bocio multinodular, hiperparatiroidismo primario y acromegalia por adenoma hipofisario. La enferma presentó, además, neoplasia de mama y pancreatitis aguda. Actualmente, para establecer el diagnóstico se acepta la asociación de 2 de los 3 hallazgos presentes en la tríada clásica, ya que en la mayoría de casos es un síndrome evolutivo y, dado que las proteínas G están ampliamente distribuidas, cualquier célula y tejido podrían verse afectadas, por lo que se podría hablar de un tumor de múltiples glándulas endocrinas.
La variedad de presentación de pacientes con SMA requiere considerar dicho diagnóstico en todos los niños con pubertad precoz atípica. También debería tenerse en cuenta a la hora de realizar un diagnóstico diferencial ante la aparición de otras endocrinopatías en la infancia. Aunque es un cuadro infrecuente, es importante la sospecha diagnóstica para poder realizar un tratamiento temprano y un cribado de las endocrinopatías asociadas al síndrome con más frecuencia, para evitar las complicaciones que se podrían derivar de una ausencia de tratamiento. Es importante no olvidar, como ya se ha comentado, que el SMA es un síndrome evolutivo, por lo que se deben realizar exploraciones periódicas, ya que las alteraciones pueden aparecer a lo largo del tiempo. Conviene detectarlas precozmente, sobre todo si existe displasia fibrosa, porque algunos de estos pacientes podrían ser portadores de un SMA no diagnosticado10. Para ello, sería útil la detección de la mutación, sobre todo, en las formas parciales y atípicas del SMA, ya que permitiría un diagnóstico temprano y un tratamiento adecuado de los niños. Sin embargo, a pesar de la elevada sensibilidad de los métodos de detección (no disponibles en todos los centros), algunas muestras pueden persistir negativas, dado que las células mutadas están confinadas únicamente a locus específicos de los tejidos afectados4. Se debe tener en cuenta, además, que la afección dependerá del número de células implicadas, por lo que no es posible predecir su importancia y su extensión. Sugerimos que se debe estudiar a estos pacientes, considerándolos de forma independiente, ya que cada uno de ellos será único. En cuanto a las implicaciones que este síndrome podría tener en la descendencia, no es necesario el consejo genético, ya que nunca es hereditario. La mutación resulta letal si se presenta en la línea germinal, y todos los descendientes son completamente normales3,4,6,9.
