


El carcinoma suprarrenal es una neoplasia poco prevalente que a menudo produce hormonas, siendo causa de síndrome de Cushing, virilización o una combinación de ambos cuadros clínicos. Más infrecuente es la aparición de un hiperaldosteronismo primario motivado por una secreción tumoral de aldosterona.
Presentamos el caso de un varón de 77 años de edad con antecedentes de hipertensión arterial de larga evolución que en las últimas semanas refería cifras elevadas de presión arterial y presentaba una hipopotasemia mantenida a pesar de tratamiento con doxazosina, diltiazem y potasio aspártico. Negaba toma de diuréticos, regaliz o esteroides fluorados. La exploración no evidenció hallazgos de interés, salvo una presión arterial de 174/86mmHg. Los resultados analíticos fueron: hemoglobina de 13g/dl (intervalo de referencia [IR]: 13–17); creatinina de 0,87mg/dl (IR: 0,70–1,22); sodio de 144mEq/l (IR: 135–148); potasio de 2,5mEq/l (IR: 3,5–5,10); coagulación y marcadores tumorales dentro de la normalidad; cortisoluria de 24h de 198μg (IR: 48–300); adrenocorticotropina de 22,7pg/ml (IR: 9–52); desoxicorticosterona de 8ng/ml (IR: 2–10); 11-desoxicortisol de 5ng/ml (IR: 0–8); dehidroepiandrosterona-sulfato de 0,5 μg/ml (IR: 2–3,3); testosterona de 4,77pg/ml (IR: 3–16); aldosterona plasmática de 66,3pg/ml (IR: 4–31); actividad renina plasmática de 0,75ng/ml/h (IR: 0,2–2,8); aldostenuria de 24h de 75μg (IR: 6–25), y cociente aldosterona/renina de 88 (IR: 0–50).
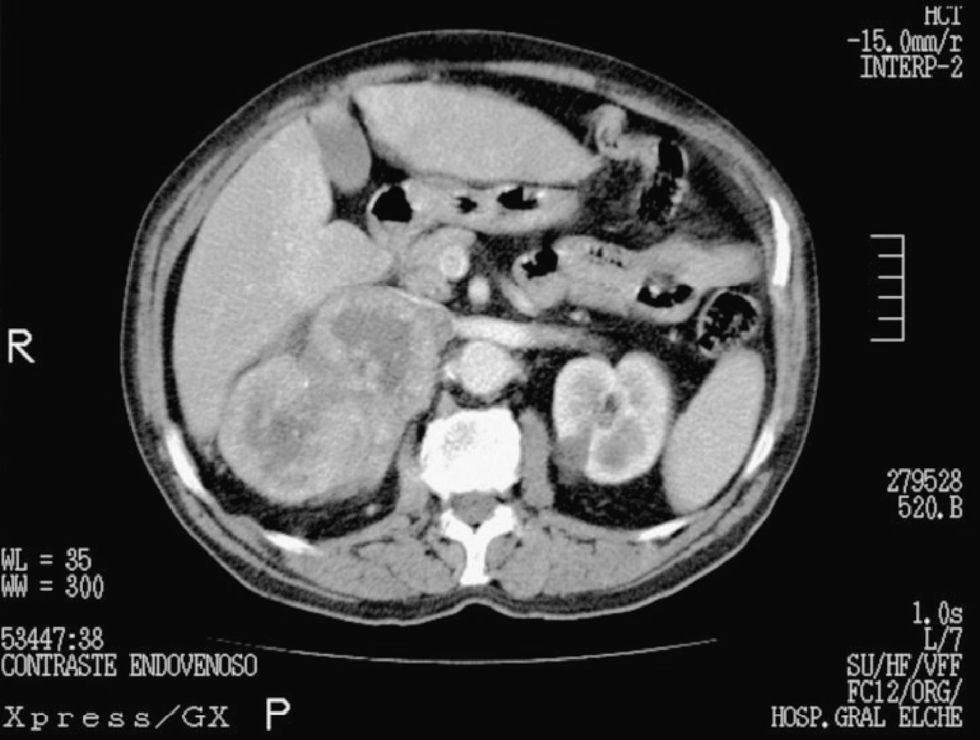
La tomografía axial computarizada (TAC) de abdomen-pelvis mostró una masa dependiente de la glándula suprarrenal derecha, polilobulada, de dimensiones 10×13×7cm, con calcificaciones en su interior y necrosis central. La lesión desplazaba el riñón derecho, la vena renal y cava inferior (fig. 1).

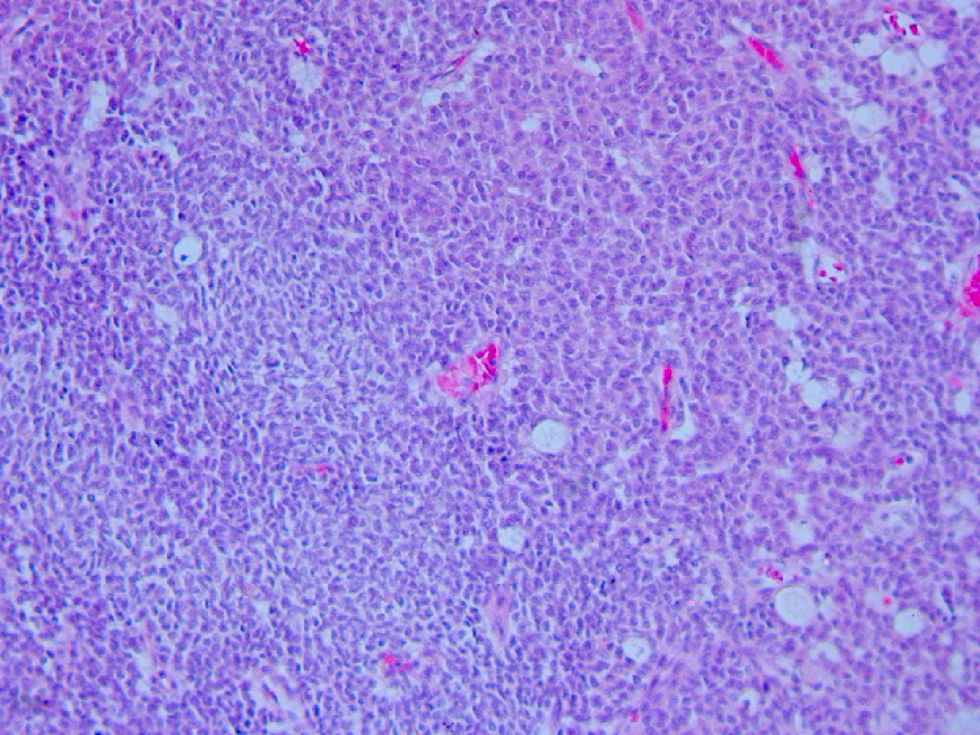
Se intentó resecar quirúrgicamente la tumoración, pero la infiltración de grandes vasos hizo imposible la exéresis, pudiéndose obtener biopsias. La tinción con hematoxilina-eosina evidenció una neoplasia de células pequeñas-medianas con moderado pleomorfismo y crecimiento difuso (fig. 2). La lesión mostraba tinción para cromogranina paranuclear y vimentina. La positividad para Ki-67 era inferior al 10%, existiendo un índice mitótico menor de 5×50 campos de gran aumento con escasas mitosis atípicas y necrosis.
 que muestra células neoplásicas de pequeño tamaño con moderado pleomorfismo y crecimiento difuso.")
El paciente rehusó cualquier terapia específica para su neoplasia, por lo que se continuó con doxazosina, iniciándose tratamiento con espironolactona (100mg/12h). Dos años después del diagnóstico, el paciente se encontró asintomático con control adecuado de presión arterial y concentraciones de potasio en límites normales. La TAC de control a los 12 meses no mostró modificaciones importantes con respecto al estudio inicial.
El carcinoma suprarrenal presenta una incidencia de un caso por millón de habitantes y año1,2, siguiendo una distribución bimodal con máximos de incidencia en la infancia y en la cuarta y la quinta décadas de la vida1. Existe un predominio femenino con una relación de 1,6:1. El 45% de las neoplasias adrenocorticales son productoras de glucocorticoides1–3, siendo entonces causa de síndrome de Cushing. Frecuentemente, estos carcinomas producen cortisol junto con otros metabolitos intermedios, fundamentalmente desoxicorticosterona y 11-desoxicortisol, que a altas concentraciones ejercen un efecto mineralocorticoide, ocasionando hipopotasemia e hipertensión arterial. El 25% de estos carcinomas puede provocar un síndrome de Cushing asociado a virilización por producción mixta de glucocorticoides y andrógenos. El 10% de los pacientes presenta virilización únicamente. Solo el 1–3% de estas neoplasias son productoras exclusivamente de aldosterona. Los casos que no asocian secreción hormonal (casi un 30%) presentan síntomas en relación con el crecimiento tumoral.
En pacientes asintomáticos debemos realizar una determinación de cortisol, estradiol, estrona, andrógenos adrenales en plasma y cortisoluria, y en aquéllos con hipopotasemia e hipertensión arterial, además, determinación de aldostenuria, aldosterona, actividad de renina y cociente aldosterona/renina en plasma. La TAC tiene alta especificidad y sensibilidad en la diferenciación entre adenomas y carcinomas. Las zonas de metástasis más frecuentes de este tumor son el hígado, los pulmones, los ganglios linfáticos y el hueso, por lo que el estudio debe ampliarse al tórax y a las regiones óseas. La resonancia magnética ofrece mayor resolución para dilucidar invasión local y afectación de la vena cava. La tomografía por emisión de positrones tras administración de fluorodeoxiglucosa nos ayuda a localizar posibles metástasis, siendo útil para discernir entre tumores benignos y malignos. Su empleo simultáneo con la TAC mejora la sensibilidad de ambas pruebas por separado en la distinción de lesiones suprarrenales4.
El sistema histopatológico de Weiss es el método más simple y reproducible para establecer la probabilidad de malignidad de una lesión adrenocortical. Dicho sistema contempla nueve criterios histológicos. La presencia de tres criterios o más en la muestra estudiada así como un tamaño tumoral superior a 5cm sugieren malignidad. Un índice muy elevado define una neoplasia de alto grado con peor pronóstico y mayor riesgo de recidiva.
La estadificación del carcinoma suprarrenal se basa en el tamaño de este, la afectación de los órganos adyacentes, la presencia de los ganglios patológicos y la metástasis. La supervivencia a los 5 años es del 66% en estadio i (tumor menor de 5cm confinado a la glándula suprarrenal), del 46% en estadio ii (tumor mayor de 5cm sin metástasis a distancia), del 21% en estadio iii (cualquier tamaño tumoral sin metástasis, pero con afectación de los tejidos adyacentes, la vena cava y/o renal y los ganglios linfáticos), y del 6% en estadio iv (tumor metastásico). La esperanza de vida de los pacientes no sometidos a ningún tipo de tratamiento oncológico no supera los 6–12 meses desde el momento del diagnóstico.
La cirugía radical es el tratamiento de elección hasta el estadio iii, siendo la única terapia que ofrece alguna posibilidad de curación, si bien la mayoría de los pacientes ya presentan enfermedad micrometastásica en el momento del procedimiento5. Durante la cirugía se debe evitar la ruptura de la cápsula tumoral, minimizando así el riesgo de diseminación intraabdominal3. Incluso cuando se realiza una cirugía con intención curativa, más del 80% de los tumores recidiva en un plazo variable de tiempo.
Respecto al tratamiento quimioterapéutico, el mitotane es un fármaco adrenocorticolítico cuyo uso en terapia adyuvante es aún muy discutido. Existe acuerdo generalizado con respecto a su uso en monoterapia en pacientes con estadios avanzados que no son candidatos a cirugía o en adyuvancia cuando tras extirpar el tumor la probabilidad de recurrencia es elevada6–8. En casos de enfermedad extendida, otros fármacos citotóxicos pueden ser combinados con mitotane. El estudio FIRM-ACT es un ensayo clínico internacional en fase iii que evalúa la eficacia de regímenes que emplean mitotane junto con otros quimioterapéuticos, como el etopósido, la doxorrubicina, el cisplatino o la estreptozotozina9. Además de un efecto citorreductor, el mitotane ofrece un importante beneficio sintomático minimizando la producción tumoral de las hormonas hasta en un 75% de los casos. Es conveniente monitorizar los niveles plasmáticos de mitotane para conseguir concentraciones terapéuticas, evitando los efectos adversos de la sobredosificación.
Se investigan nuevas terapias basadas en el empleo de anticuerpos contra los receptores del factor de crecimiento insulínico tipo i, inhibidores de la tirosina-kinasa y fármacos antiangiogénicos9. Sin embargo, la experiencia se reduce a casos aislados con resultados variables.
El papel de la radioterapia es controvertido, aunque parece ser de gran utilidad en adyuvancia tras la cirugía, previniendo las recidivas locales en pacientes de alto riesgo. También tiene indicación como tratamiento paliativo, consiguiendo importante mejoría sintomática10.
Como se ha comentado, es excepcional que un carcinoma suprarrenal produzca exclusivamente aldosterona, aunque lo realmente llamativo de este caso es la supervivencia que presentó el paciente. Pensamos que la clave de un curso clínico tan inusual radica en la propia biología del tumor. A pesar del gran tamaño de la lesión y la infiltración de grandes vasos, la histología evidenciaba un bajo índice mitótico con escasas atipias y un grado nuclear moderado. Ello indica que estamos ante un tumor de crecimiento lento y poco agresivo.
