


La apoplejía hipofisaria se define como el síndrome clínico que se da tras la hemorragia o infarto espontáneo de la glándula pituitaria, en general en el contexto de un adenoma preexistente1,2. Su etiopatogenia no se conoce con exactitud, aunque existen múltiples factores de riesgo descritos y parece que es más frecuente en macroadenomas no funcionantes2. Clínicamente se manifiesta como cefalea brusca, acompañada de náuseas y vómitos, alteraciones visuales, hipopituitarismo y disminución del nivel de conciencia llegando incluso a un estado de coma. El tratamiento es inicialmente de soporte, siendo necesaria en algunos casos la descompresión quirúrgica, sobre todo si existe compromiso visual3. Tras el episodio, todos los pacientes requieren un seguimiento a largo plazo ya que la presencia de una disfunción hipofisaria residual y/o reaparición o persistencia de un tumor preexistente no es rara4–6. Los casos descritos en enfermedad de Cushing son escasos4,5. A continuación presentamos un caso de curación espontánea de enfermedad de Cushing tras un episodio de apoplejía hipofisaria.
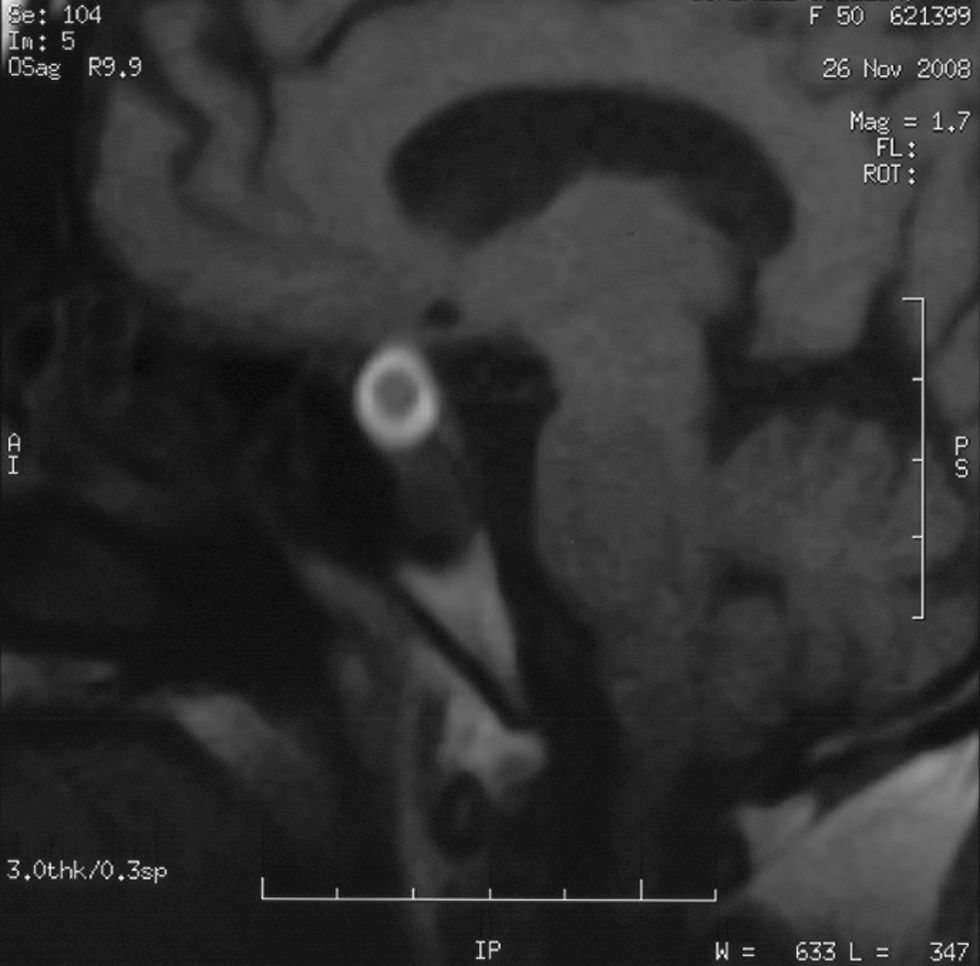
Mujer de 50 años, con antecedentes de HTA y trastorno bipolar, que es remitida por sospecha de síndrome de Cushing. A la exploración física destacaba obesidad de predominio central, con estrías rosadas en abdomen, cara de luna llena y rubicundez facial. Presentaba giba y ocupación grasa de fosas supraclaviculares, así como hirsutismo y múltiples hematomas, junto con amiotrofia de extremidades inferiores. Refería amenorrea secundaria desde hacía 8 meses, con oligomenorrea en los últimos 5 años. El estudio hormonal mostró cortisoluria elevada: 787,8μg/24h (55–325), cortisol basal de 45,99μg/dl (4,30–24,40) y ACTH de 170pg/ml (9–52). Presentaba también hipogonadismo hipogonadotropo, siendo el resto de la función hipofisaria normal. Se realizó supresión débil con 2mg de dexametasona obteniéndose un cortisol de 30,85μg/dl, y supresión fuerte con 8mg de dexametasona con cortisol de 6,12μg/dl. Dos semanas después del diagnóstico la paciente consultó por hipotensión arterial y empeoramiento del estado general, refiriendo astenia, náuseas y vómitos y un episodio de cefalea holocraneal opresiva 10 días antes. La RM hipofisaria mostró una tumoración intraselar de 1,3×1,4×1,5cm, sin invasión del seno cavernoso y con crecimiento supradiafragmático, con anillo a su alrededor engrosado en T1 compatible con una hemorragia subaguda (fig. 1). Con la sospecha de insuficiencia suprarrenal aguda secundaria a una apoplejía hipofisaria, se inició tratamiento con hidrocortisona endovenosa y sueroterapia, con mejoría de las cifras tensionales y del estado general. Se realizó una campimetría que resultó ser normal. Las concentraciones de ACTH fueron de 33,2pg/ml, con cortisoluria de 259,8μg/24h. En las analíticas realizadas posteriormente la paciente ha presentado remisión del hipercortisolismo (cortisoluria 121μg/24h, cortisol basal 18,1μg/dl, ACTH 24,8pg/ml, cortisol tras frenación con 2mg de dexametasona: 1,12μg/dl).

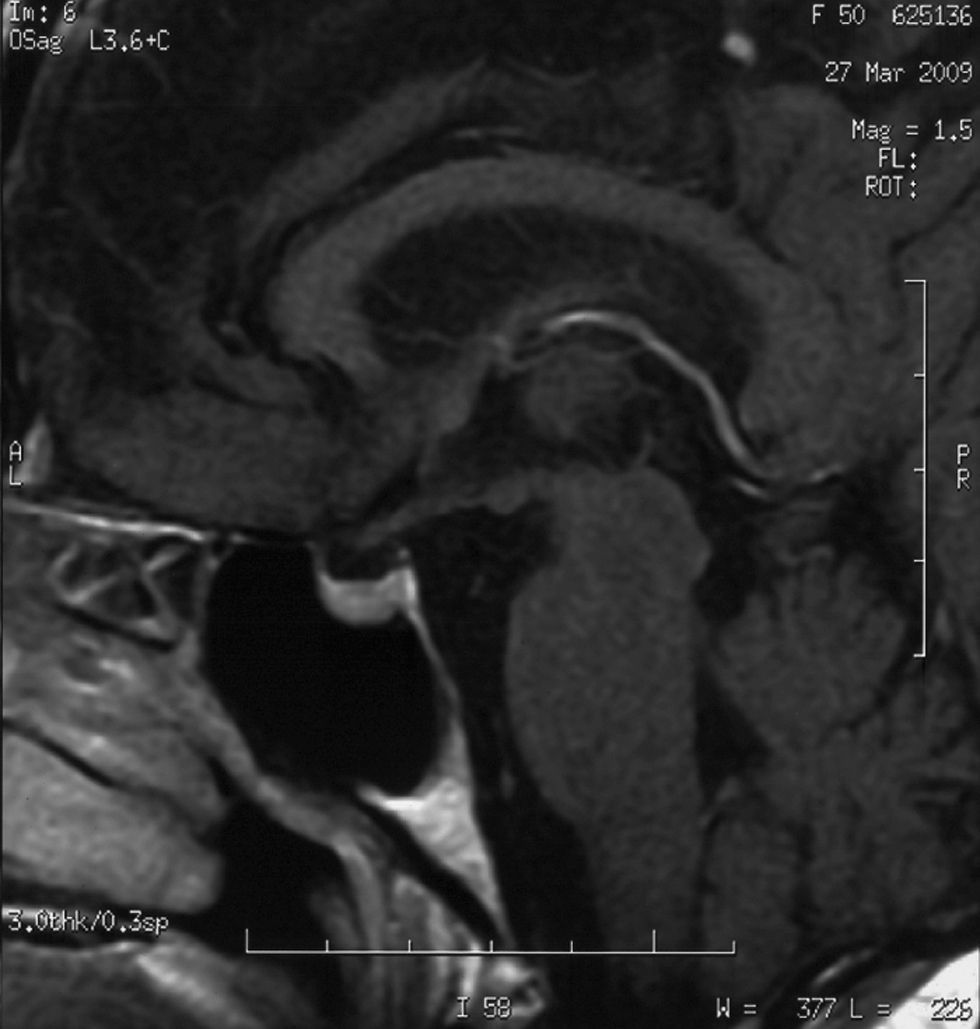
La RM realizada 3 meses después (fig. 2) mostró la desaparición de la tumoración detectada en la exploración previa. Persistía cierta desviación del tallo hipofisario a la izquierda, siendo el resto normal. En el seguimiento, las concentraciones de cortisoluria se mantienen en niveles normales y el resto de la función hipofisaria está conservada.

Sin embargo, la disfunción parcial o total de la hipófisis anterior tras el episodio es frecuente, produciéndose una destrucción selectiva del tumor con conservación de la función hipofisaria solo en casos aislados6.
El seguimiento a largo plazo es importante ya que la recidiva de la enfermedad de Cushing tras un episodio de apoplejía hipofisaria puede ocurrir incluso varios años después del episodio4.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
