


La diabetes sintomática como manifestación de otro proceso subyacente es un fenómeno infrecuente, y usualmente obedece al empleo de fármacos con potencial hiperglucemiante, o a procesos neoplásicos del área pancreática. Entre los fármacos, destacan por frecuencia los esteroides a dosis elevadas, y, más recientemente y en pacientes jóvenes, los neurolépticos atípicos1. El debut cardinal de una diabetes mellitus (DM) en un paciente menor de 40 años, sin otra clínica organotópica ni tratamiento concomitante orienta a una diabetes autoinmune (tipo 1A) como primera posibilidad, mientras que en ancianos obliga a descartar un proceso neoplásico subyacente. Aportamos el caso de un aparente debut de DM tipo 1 cuya evaluación posterior demostró un diagnóstico etiológico inesperado y reversible.
Un varón de 38 años, sin antecedentes personales ni familiares de interés, ingresó procedente de urgencias por hiperglucemia con cetosis. El paciente refería un cuadro de poliuria y polidipsia intensa de 3 semanas de evolución, acompañado de la pérdida de 12Kg en el último año sin disminución de apetito ni otra sintomatología añadida. En el servicio de urgencias destacaba una glucemia 340mg/dl, cuerpos cetónicos en orina de 50mg/dl y pH 7,4. El peso era de 56,5Kg (IMC: 17Kg/m2), tensión arterial 145/95mmHg y frecuencia cardíaca de 100 latidos por minuto, sin otros hallazgos en la exploración física. Se pautó insulinoterapia en régimen basal-bolus (glargina y aspart), y educación diabetológica, consiguiendo un buen control glucémico. La hemoglobina glucosilada al debut fue de 13,4%, con anticuerpos anti-IA2 (insulinoma antigen 2) y anti-GAD (descarboxilasa del ácido glutámico) negativos, y un péptido C basal de 2,2ng/ml, por lo que se desestimó la posibilidad de una DM tipo 1A, y se prosiguió el estudio para descartar causas secundarias.
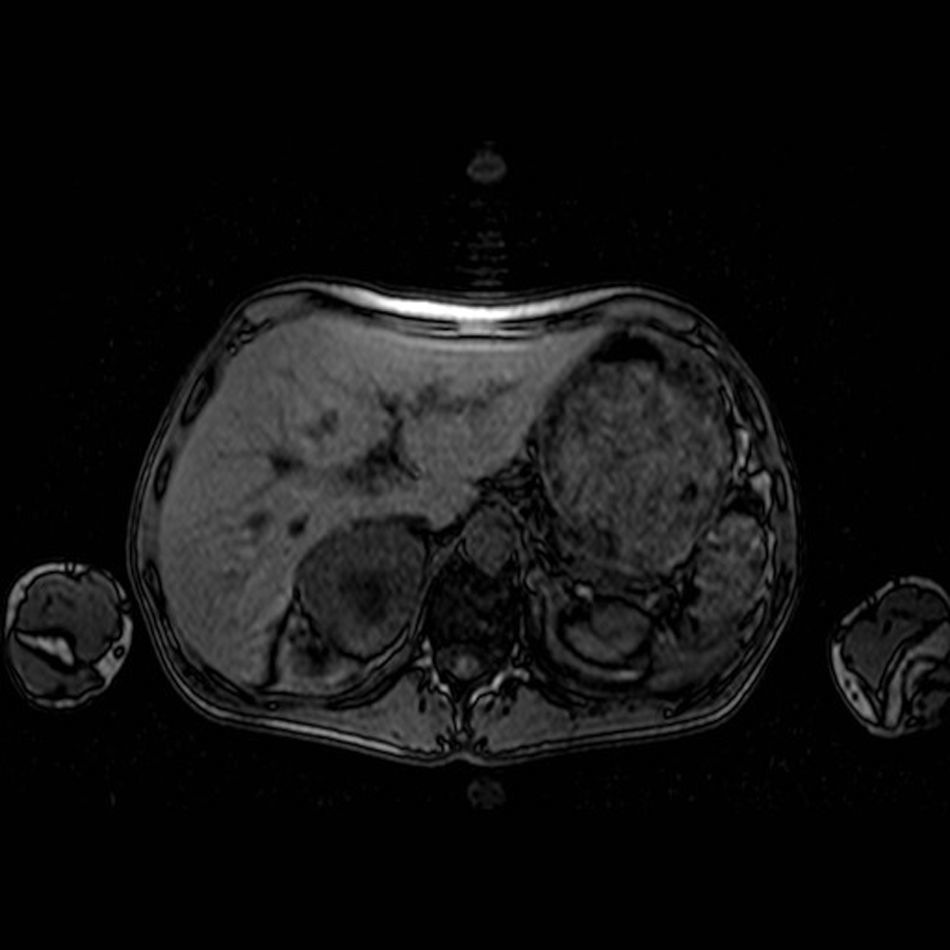
Las hormonas tiroideas, cortisol, calcitonina, calcio y fósforo eran normales. Una ecografía abdominal objetivó una masa suprarrenal derecha de 7cm, que en la resonancia magnética se confirmó como una tumoración heterogénea con centro necrótico (fig. 1). La medición de catecolaminas y sus metabolitos en orina de 24 horas arrojaron los siguientes resultados: norepinefrina 767μg/24h (normal [N]: 12,1-85,5), epinefrina 270μg/24h (N: 1,7-22,4), dopamina 461μg/24h (N: 0-498), normetanefrinas 3.245μg/24h (N: 88-444) y metanefrinas 2.299μg/24h (N: 52-341). Tras la preparación prequirúrgica con fenoxibenzamina, sin precisar betabloqueantes, a las 4 semanas se realizó una suprarrenalectomía laparoscópica sin incidencias. El estudio anatomopatológico confirmó un feocromocitoma de 7,5×5 x 5cm y 138g de peso, sin invasión capsular. Tras la cirugía las necesidades de insulina disminuyeron de forma llamativa y a los pocos días no precisaba tratamiento hipoglucemiante. En revisiones posteriores hasta la actualidad, el paciente ha persistido asintomático, euglucémico y normotenso, ha recuperado su peso habitual y las determinaciones hormonales han permanecido normales.

El feocromocitoma es un tumor productor de catecolaminas derivado de las células enterocromafines de la médula adrenal. Es un tumor poco frecuente, diagnosticándose aproximadamente 1-2 casos por 100.000 habitantes y año2, aunque su prevalencia es mayor (0,05-0,1%) en estudios post mortem3,4. La presentación clínica puede ser muy variada, desde pacientes asintomáticos hasta crisis hipertensivas severas5. Aunque la triada clásica de palpitaciones, cefalea y sudoración es muy específica del diagnóstico, la mayoría de los pacientes presentan hipertensión arterial mantenida o paroxística5 sin otro correlato clínico. La prevalencia de algún tipo de alteración del metabolismo hidrocarbonado en los casos de feocromocitoma es muy variable según distintos estudios. En una serie de 60 pacientes, el 24% presentó DM y se observó una clara relación entre los niveles de catecolaminas urinarias y las cifras de glucemia6. En otra serie de 191 pacientes la prevalencia fue algo mayor del 35%7. En cambio, en un estudio realizado a 1.093 pacientes diabéticos con ecografía abdominal y prueba de glucagón se diagnosticó de feocromocitoma a 0,96/1.000 pacientes8, prevalencia similar a la encontrada en población hipertensa (1/1.000)2. A pesar de que la hiperglucemia secundaria a feocromocitoma no sea un hecho infrecuente, es raro el debut con clínica cardinal, y existen solamente 4 casos descritos en la literatura con cetoacidosis9. En nuestro caso, aunque no presentó acidosis, la hiperglucemia y la cetosis eran muy importantes y dada la edad del paciente sugería un debut de diabetes tipo 1 clásico. La ausencia de clínica típica de feocromocitoma, aparte de la hipertensión, demoró el diagnóstico final.
En el feocromocitoma, la alteración hidrocarbonada es secundaria a múltiples factores. Por un lado, existe una disminución de la secreción de insulina, aumento de las concentraciones de glucagón y estimulación de la glucogenólisis secundaria al aumento de norepinefrina y, por otro lado, una disminución de la captación periférica de glucosa y aumento de la gluconeogénesis hepática secundaria al exceso de epinefrina. Tras la exéresis del tumor la mayoría de los pacientes no precisan ningún tratamiento hipoglucemiante10.
En conclusión, destacamos la variabilidad en la forma de presentación de estos tumores, en este caso únicamente manifiesto por la progresiva pérdida ponderal hasta el debut cardinal de la diabetes mellitus. Por ello, debemos tener presente la posibilidad de un feocromocitoma en pacientes jóvenes con hipertensión arterial y diabetes mellitus no autoinmune7,8.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
