


La deficiencia de tirotropina (TSH) como origen del hipotiroidismo constituye una excepción en la práctica clínica y, en la mayoría de los casos, se enmarca en un déficit antehipofisario múltiple. Este defecto plurihormonal es usualmente secundario a una lesión estructural, tumoral o vascular, o bien se debe al tratamiento de un proceso selar o paraselar. La deficiencia aislada de TSH se identifica en la infancia en portadores de mutaciones en la subunidad β de la TSH1 o en el receptor de la hormona estimulante de la secreción de TSH (TRH)2. Su detección aislada en la vida adulta constituye un enigma para el clínico, particularmente al tratar de establecer su etiología.
Detallamos el caso de un paciente diagnosticado de déficit aislado de TSH en la edad adulta y planteamos su posible etiología sobre la base de los hallazgos clínicos, analíticos y radiológicos.
Un varón de 46 años sin antecedentes relevantes fue remitido al encontrársele una tiroxina libre (T4L) baja con TSH normal en una analítica de control. Retrospectivamente, refería astenia moderada que atribuía a la sobrecarga laboral, sin variaciones en el peso ni el ritmo intestinal. No presentaba síntomas de disfunción adrenal ni hipogonadismo y negaba el consumo reciente de fármacos. En sendos análisis distanciados por una semana se evidenciaban T4L de 5,9–6,0pmol/l (valores normales: 12–26), con TSH simultáneas normales (2,54–2,66mUI/l), anticuerpos antiperoxidasa tiroidea negativos y dislipemia de fenotipo IIA (colesterol de las lipoproteínas de baja densidad: 197 y 190mg/dl), sin anemia ni alteraciones iónicas. Su fenotipo era normal, con talla acorde a las tallas familiares, y tensión arterial normal.
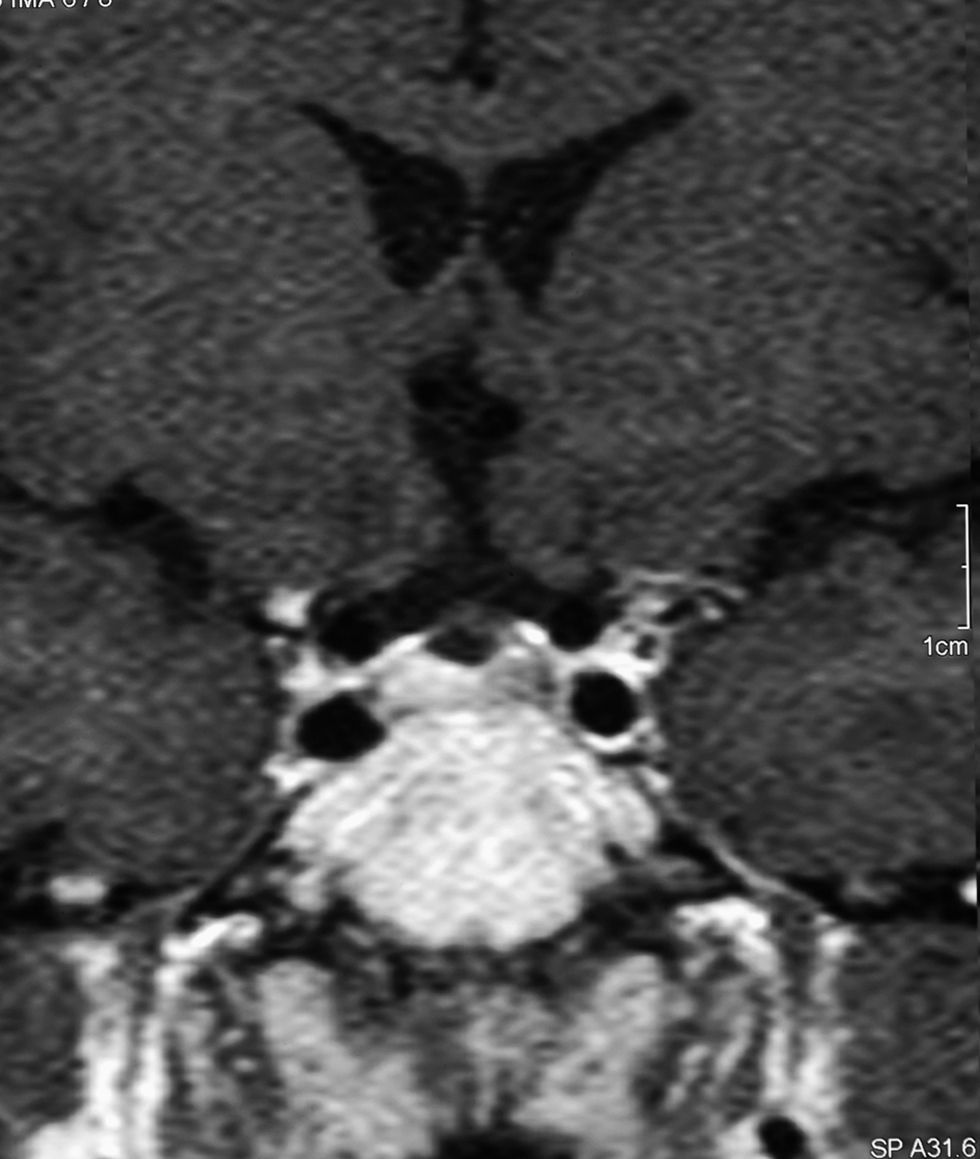
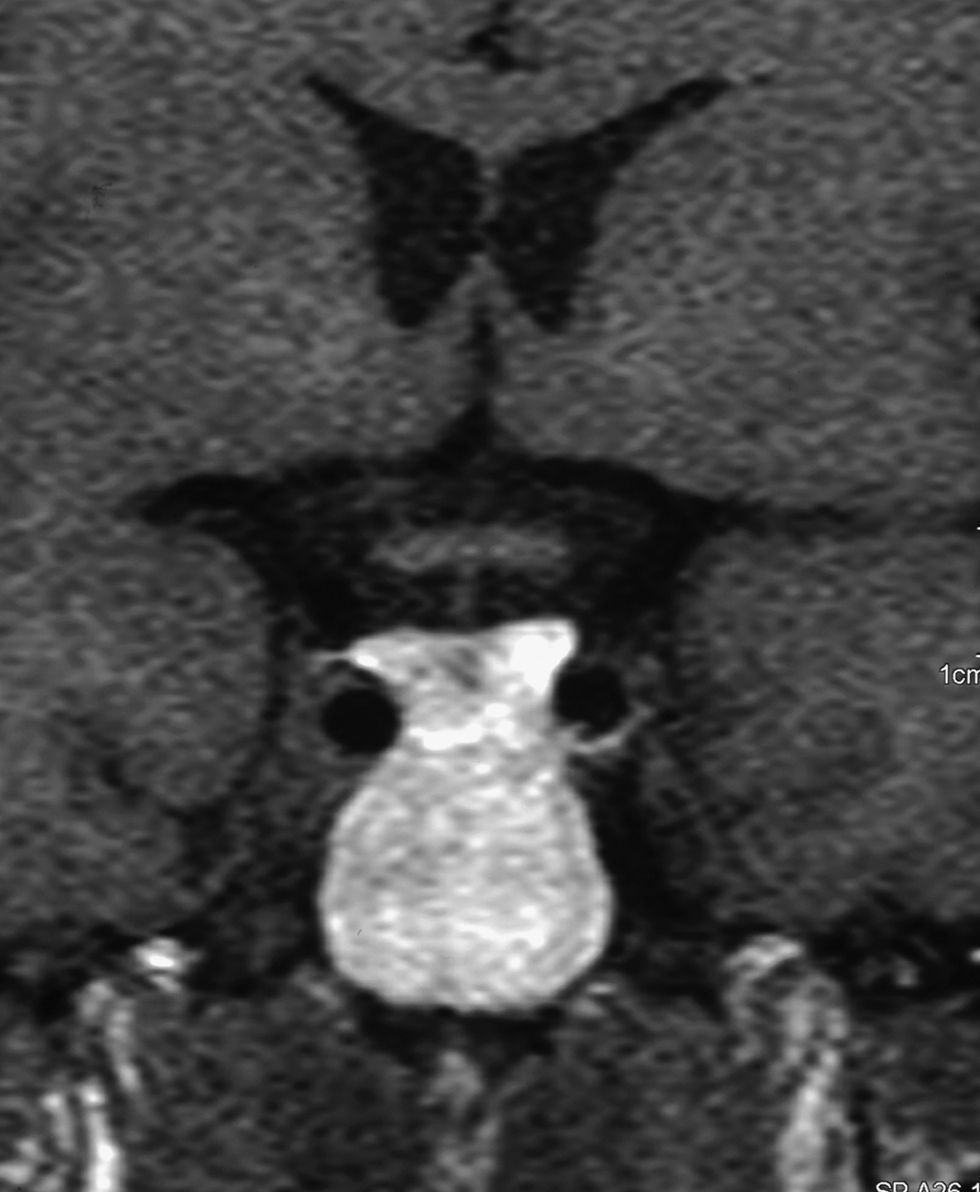
Tras pautar L-tiroxina con cobertura esteroidea, el estudio de función hipofisaria descartó la afectación de otros ejes (cortisol tras 250μg de adrenocorticotropina endovenosa: 27,2μg/dl; testosterona: 5,57ng/ml; hormona luteotropa y foliculoestimulante: 4,6 y 10,2UI/l, respectivamente; prolactina: 12μg/l, y somatomedina-C: 131ng/ml [VN: 109–238]). Tras retirar la terapia seis semanas, se reevaluó al paciente con un test de TRH, con concentración basal de 2,23 y pico de 3,28mUI/l a los 20min del estímulo. Una resonancia magnética (RMN) selar evidenció una lesión hipofisaria hipodensa de 4mm lateral izquierda, mal definida y no captante, y con el tallo hipofisario desviado a la derecha (figs. 1 y 2). Tras reiniciar L-tiroxina, a los 3 meses refería disminución de la astenia, mayor temperatura corporal y aumento de la frecuencia deposicional. La cifra de colesterol de lipoproteínas de baja densidad se redujo hasta 120mg/dl. En los sucesivos controles se ha mantenido asintomático, con niveles de testosterona, cortisol e IGF-I normales. En la RMN, pasados 12 meses, persistía la imagen inicial. Reinterrogado, no aportaba datos previos de interés y en el archivo histórico se localizó una analítica, tres años antes de la primera consulta, en la que aparecía la dislipemia que respondió al tratamiento.


El hipotiroidismo central, definido como una T4L plasmática baja con valores normales de TSH, tiene una prevalencia estimada del 0,005% de la población general frente a la elevada prevalencia del hipotiroidismo primario. En su presentación más habitual forma parte de un hipopituitarismo de origen tumoral, infiltrativo, traumático, isquémico o en relación con el tratamiento quirúrgico o la radioterapia del área selar. Los casos congénitos se identifican en presencia de mutaciones de genes homeobox implicados en el desarrollo hipofisario (HESX1, LHX3, PROP1 y POU1F1), con fenotipos variables por la presencia de diversos déficits simultáneos. La deficiencia aislada es excepcional y aparece en la mutación de la subunidad β de la TSH1, o en la inactivación del receptor de TSH por mutaciones heterocigotas compuestas2, esta última de diagnóstico tardío en la infancia. En los déficits aislados de la vida adulta se han propuesto como causas potenciales un aumento del tono dopaminérgico o somatostatinérgico, e incluso una mayor actividad de la actividad opioide cerebral. Por este mecanismo, la administración aguda de dopamina, somatostatina, dobutamina y glucocorticoides produce una inhibición transitoria en la TRH, pero sin repercusión clínica. La terapia con bexaroteno, agonista del receptor del retinoide X empleado en el tratamiento del linfoma de células T, produce un hipotiroidismo de patrón central a través de la inhibición del promotor de la subunidad β de la TSH3.
Clásicamente se defendía la utilidad del estímulo con TRH para definir el nivel de alteración, con respuesta tardía indicativa de un origen hipotalámico y con respuesta plana en el hipofisario4. Hoy en día, esta distinción ha perdido valor al disponer de técnicas de radiodiagnóstico que permiten un diagnóstico anatómico preciso en la mayoría de los casos.
Característicamente, la cifra de TSH en el hipotiroidismo central permanece detectable, incluso en el límite alto, a diferencia de otras deficiencias hipofisarias. La persistente secreción de una TSH inmunorreactiva, pero bioinactiva debido a un patrón de glucosilación anómalo de la molécula5, explica este fenómeno. La expresividad clínica del hipotiroidismo central es menos florida que la del primario, con niveles bajos pero detectables de hormonas tiroideas, que atenúan la expresión clínica. La causa radica en que una pequeña fracción de la función tiroidea, en torno al 10–15%, es independiente de la TSH, por lo que el hipotiroidismo central es menos severo que el primario6.
El paciente descrito cumplía todos los criterios de una deficiencia aislada de TSH, de aparente origen hipofisario, con la particularidad de haberse iniciado en la vida adulta y no cursar con daño estructural selar, por lo que su etiología resultaba incierta. Pese a su escasa sintomatología inicial, reafirmaban la presencia de un hipotiroidismo clínico de larga data y descartaban un posible síndrome de tiroides enfermo eutiroideo. El papel potencial de neurotransmisores o fármacos inhibidores de la TRH parece improbable, ya que el paciente no había tomado medicación alguna.
La deficiencia aislada de otras hormonas de la adenohipófisis es también inusual si exceptuamos los déficits de hormona de crecimiento y/o gonadotropinas en lesiones hipofisarias parciales. El déficit aislado de adrenocorticotropina ha sido comunicado en el seno de los fenómenos autoinmunes7, pero hasta la fecha no se ha publicado la conjunción de hipofisitis y déficit aislado de TSH, y la RMN tampoco avalaba esta posibilidad.
En pacientes con tumores somatotropos, prolactinomas y en adenomas corticotropos se ha descrito la curación espontánea por necrosis del tumor, incluso con déficits posteriores del factor previamente hipersecretado8,9. Asimismo, los pacientes intervenidos de un microadenoma productor de TSH pueden presentar un patrón de hipotiroidismo central postoperatorio de duración variable que puede hacer necesario el empleo de terapia sustitutiva10, similar a la insuficiencia suprarrenal que sucede a la curación quirúrgica de una enfermedad de Cushing, y que puede permanecer de forma indefinida.
La imagen obtenida en este paciente, compatible con un microadenoma posiblemente infartado, junto con su cuadro clínico, nos sugiere la posibilidad de un déficit aislado de TSH acaecido tras la lesión espontánea y resolución de un adenoma productor de TSH, bien silente, o bien sin apenas expresión clínica. La comprobación de esta hipótesis demandaría la intervención quirúrgica y obtención del tumor, pero la excelente respuesta al tratamiento y su estabilidad radiológica y hormonal no avalan dicha actitud. Esta secuencia potencial no aparece reflejada en la literatura médica, pero el carácter adquirido y permanente del déficit, junto con el correlato radiológico, permiten considerarla una, sino la única, etiología posible.
